
Reversible Chemical Modification Strategies and Applications in Protein Chemical Synthesis
† These author contributed equally to this work
Received date: 2024-09-09
Revised date: 2024-10-15
Online published: 2025-06-17
Supported by
National Natural Science Foundation of China(22277020)
National Natural Science Foundation of China(22227810)
National Natural Science Foundation of China(22207001)
Natural Science Foundation of Anhui Province(2208085QC74)
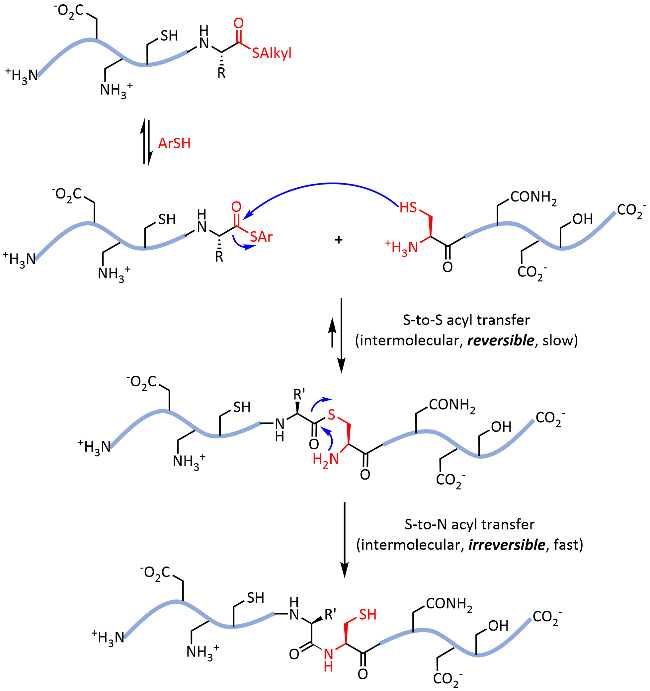
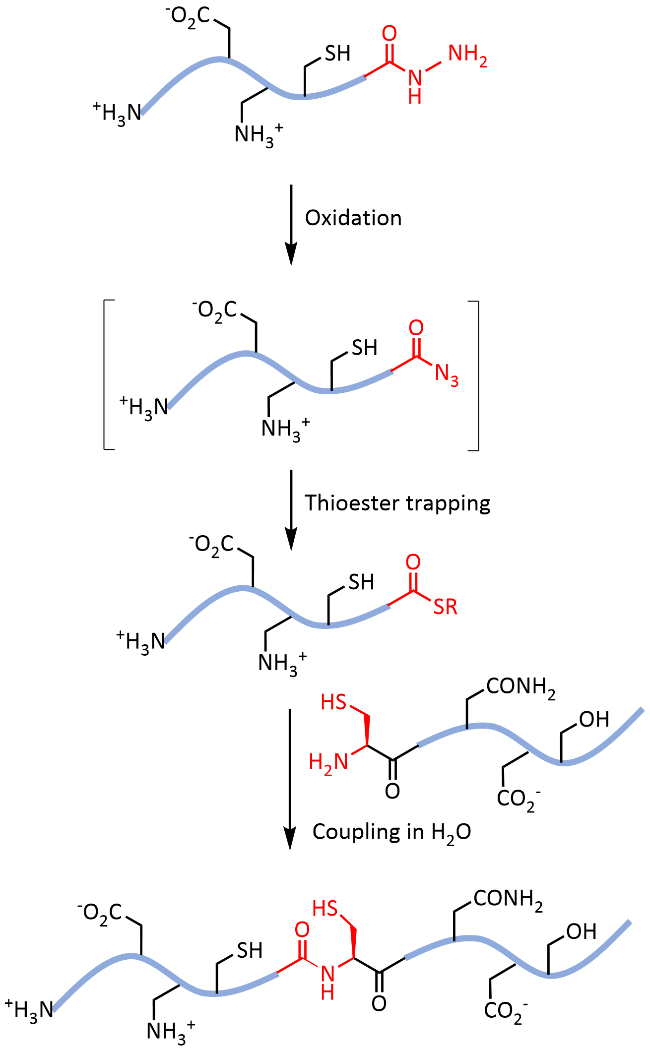
Protein chemical synthesis plays a crucial role in preparing proteins with specific sequences. Although this technology has been successfully applied to the synthesis of various proteins, the issues of solubility and refolding efficiency remain significant challenges for researchers when synthesizing hydrophobic and disulfide-rich proteins. The introduction of reversible chemical modification tags to the side chains or backbone of proteins offers an effective solution. Specifically, the introduction of solubilizing-tag during the protein synthesis process can significantly improve the water solubility of hydrophobic peptide segments, thereby facilitating subsequent protein synthesis and purification. The introduction of glycosylation modification effectively improves the folding of disulfide-rich proteins by stabilization of their folding intermediates. Moreover, these reversible modification tags can ultimately be removed by specific chemical or biological conditions, ensuring that the biological activity and structural integrity of the proteins are unaffected. This review delves into the types, introduction strategies and removal conditions of reversible modification tags and details their important applications in protein synthesis. These strategies not only expand the tools of protein chemical synthesis but also provide strong support for biomedical research and drug development, promising to drive further development in related fields.
1 Introduction
2 Reversible chemical modification strategies for synthesizing hydrophobic proteins
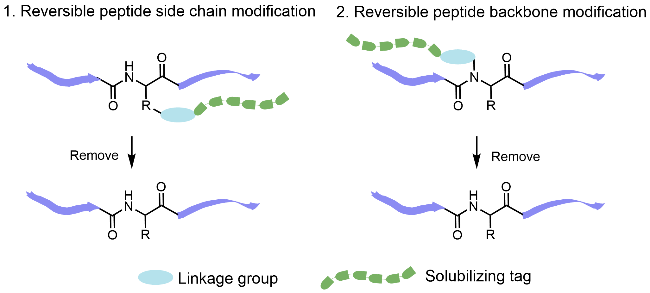
2.1 Introduction of reversible modification tags in peptide side chains
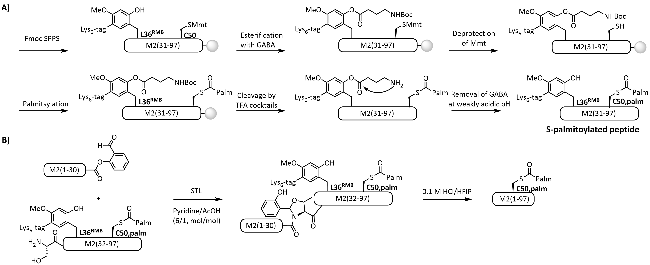
2.2 Peptide backbone modification
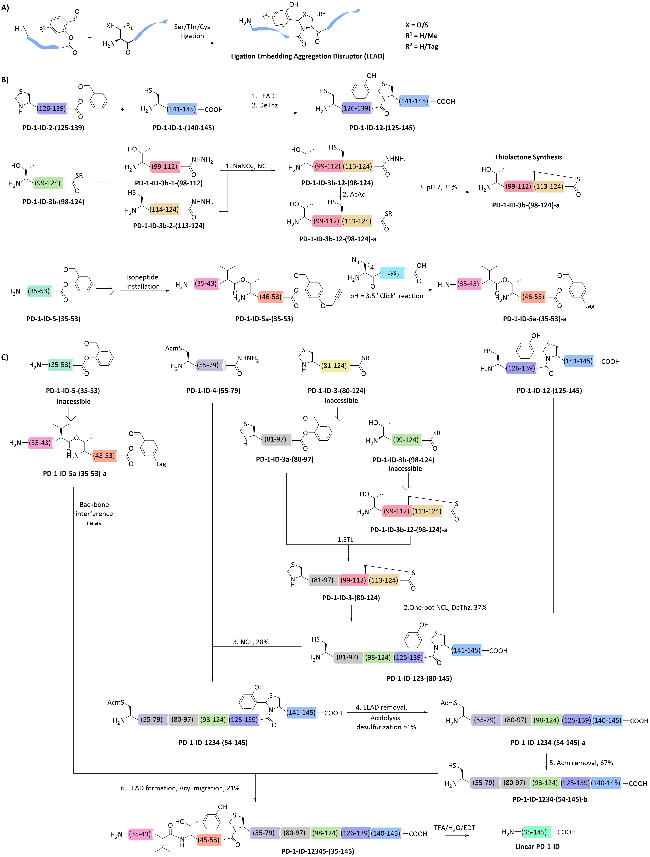
3 Reversible glycosylation modification strategies for the synthesis of difficult-to-fold proteins
4 Conclusion and outlook
Ling Xu , Tingting Cui , Yiming Li . Reversible Chemical Modification Strategies and Applications in Protein Chemical Synthesis[J]. Progress in Chemistry, 2025 , 37(6) : 882 -902 . DOI: 10.7536/PC240818
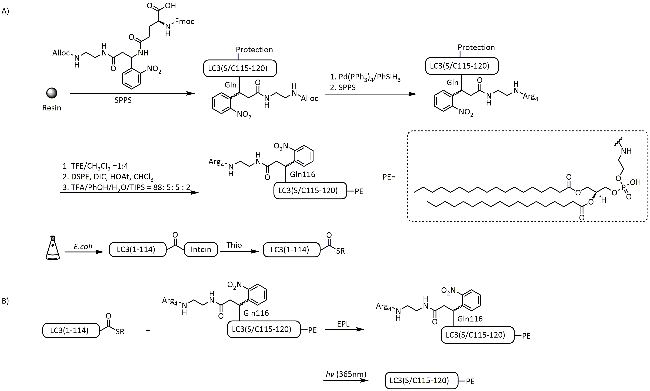
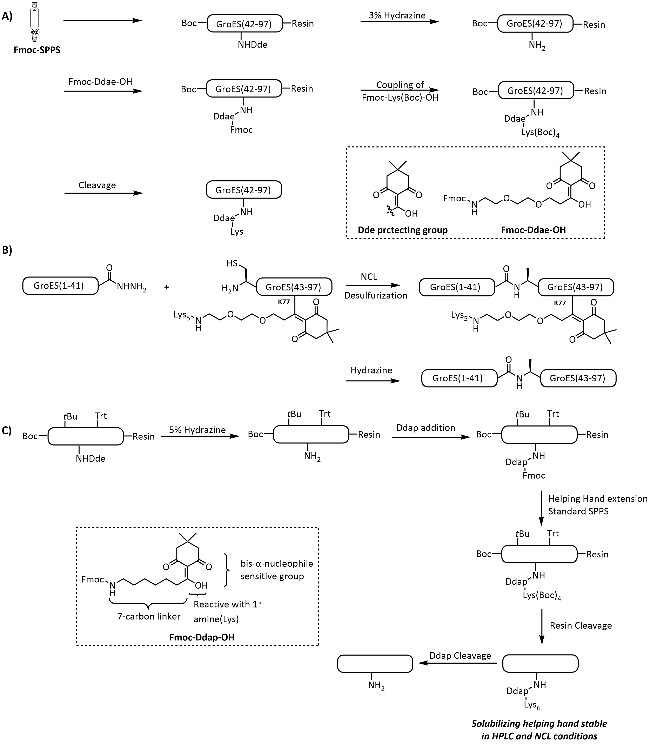
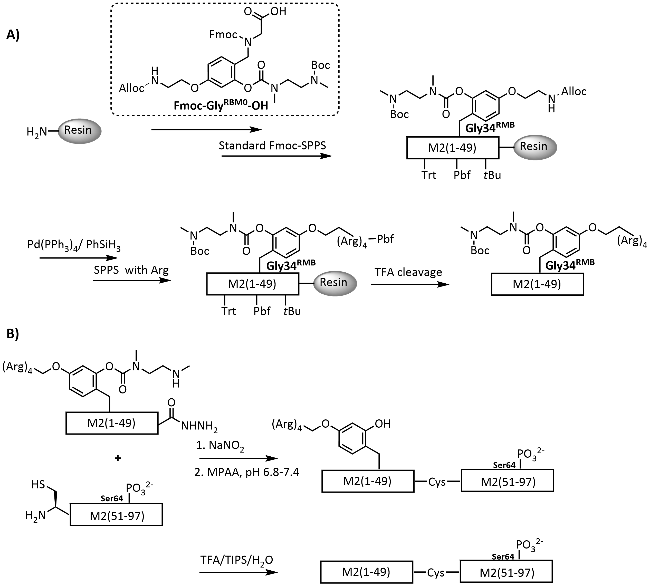
图式10 通过Ddae/Ddap连接体引入助溶标签以合成目标蛋白[111-112]: (A) Ddae连接体的引入策略;(B) 基于酰肼的自然化学连接合成GroES蛋白; (C) Ddap连接体的引入策略Scheme 10 Introduction of solubilizing tag via Ddae/Ddap linker for synthesis of target proteins[111-112]. (A) Strategies for the introduction of Ddae linker to peptide fragments; (B) Hydrazine-based native chemical ligation for synthesis of GroES; (C) Strategies for the introduction of Ddap linker to peptide fragments |
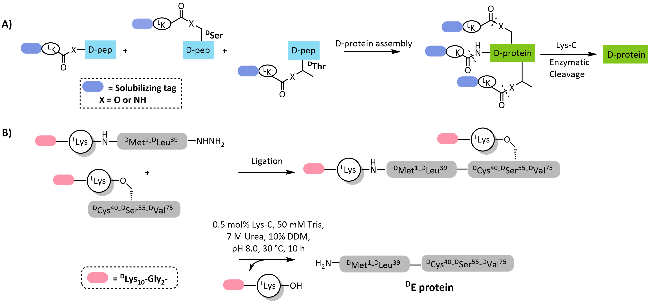
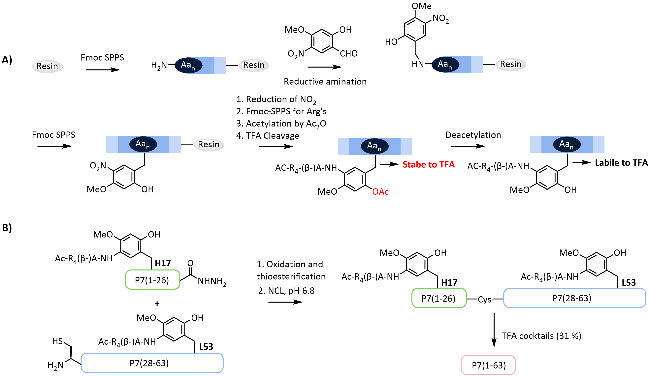
图式11 L-Lys连接的助溶标签用于合成DE蛋白[113]: (A) L-Lys连接的助溶标签的引入策略;(B) 基于酰肼的自然化学连接合成DE蛋白Scheme 11 Chemical synthesis of DE protein via the L-Lys linked solubilizing tag[113]. (A) Strategies for the introduction of L-Lys linked solubilizing tag; (B) Hydrazine-based native chemical ligation for synthesis of DE protein |
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|
| [24] |
|
| [25] |
|
| [26] |
|
| [27] |
|
| [28] |
|
| [29] |
|
| [30] |
|
| [31] |
|
| [32] |
|
| [33] |
|
| [34] |
|
| [35] |
|
| [36] |
|
| [37] |
|
| [38] |
|
| [39] |
|
| [40] |
|
| [41] |
|
| [42] |
|
| [43] |
|
| [44] |
|
| [45] |
|
| [46] |
|
| [47] |
|
| [48] |
|
| [49] |
|
| [50] |
|
| [51] |
|
| [52] |
|
| [53] |
|
| [54] |
|
| [55] |
|
| [56] |
|
| [57] |
|
| [58] |
|
| [59] |
|
| [60] |
|
| [61] |
|
| [62] |
|
| [63] |
|
| [64] |
|
| [65] |
|
| [66] |
|
| [67] |
|
| [68] |
|
| [69] |
|
| [70] |
|
| [71] |
|
| [72] |
|
| [73] |
|
| [74] |
|
| [75] |
|
| [76] |
|
| [77] |
|
| [78] |
Gluhacevic von Krüchten D,
|
| [79] |
|
| [80] |
|
| [81] |
|
| [82] |
|
| [83] |
|
| [84] |
|
| [85] |
|
| [86] |
|
| [87] |
|
| [88] |
|
| [89] |
|
| [90] |
|
| [91] |
|
| [92] |
|
| [93] |
|
| [94] |
|
| [95] |
|
| [96] |
|
| [97] |
|
| [98] |
|
| [99] |
|
| [100] |
|
| [101] |
|
| [102] |
|
| [103] |
|
| [104] |
|
| [105] |
|
| [106] |
|
| [107] |
|
| [108] |
|
| [109] |
|
| [110] |
|
| [111] |
|
| [112] |
|
| [113] |
|
| [114] |
|
| [115] |
|
| [116] |
|
| [117] |
|
| [118] |
|
| [119] |
|
| [120] |
|
| [121] |
|
| [122] |
|
| [123] |
|
| [124] |
|
| [125] |
|
| [126] |
|
| [127] |
|
| [128] |
|
| [129] |
|
| [130] |
|
| [131] |
|
| [132] |
|
| [133] |
|
| [134] |
|
| [135] |
|
| [136] |
|
| [137] |
|
| [138] |
|
| [139] |
|
| [140] |
|
| [141] |
|
| [142] |
|
| [143] |
|
| [144] |
|
| [145] |
|
| [146] |
|
| [147] |
|
| [148] |
|
| [149] |
|
| [150] |
|
| [151] |
|
| [152] |
|
| [153] |
|
| [154] |
|
| [155] |
|
| [156] |
|
| [157] |
|
| [158] |
|
| [159] |
|
| [160] |
|
| [161] |
|
| [162] |
|
| [163] |
|
| [164] |
|
| [165] |
|
| [166] |
|
| [167] |
|
| [168] |
|
| [169] |
|
/
| 〈 |
|
〉 |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}