
Oxygen Storage and Release Mechanism of Oxygen Carriers
Received date: 2024-12-24
Revised date: 2025-03-15
Online published: 2025-08-05
Supported by
the National Natural Science Foundation of China(52174279)
Chemical looping (CL) technology has been widely used in fields such as in-situ capture of carbon dioxide,hydrogen production,oxidative dehydrogenation and partial oxidation of methane. The development of oxygen carriers is the key link to the advancement of CL. Exploring the mechanism of oxygen storage and release in the oxygen carrier lattice is important for the design of high-performance oxygen carriers,the explanation of CL reaction mechanism,and the regulation of product selectivity and yield. First,this paper systematically reviews the research methods and progress of oxygen storage and release mechanism of oxygen carriers,presenting the important role of key characterization techniques in exploring the lattice oxygen migration mechanism. At the same time,we summarize the reaction mechanism of different types of oxygen carriers and the spatiotemporal evolution characteristics of active components,providing theoretical support for the design and modification of oxygen carriers. Furthermore,this paper also focuses on the difficulties and controversies in the study of oxygen storage and release mechanism of CL oxygen carriers. Finally,some perspectives on the current studies of mechanism for oxygen carriers were presented.
1 Introduction
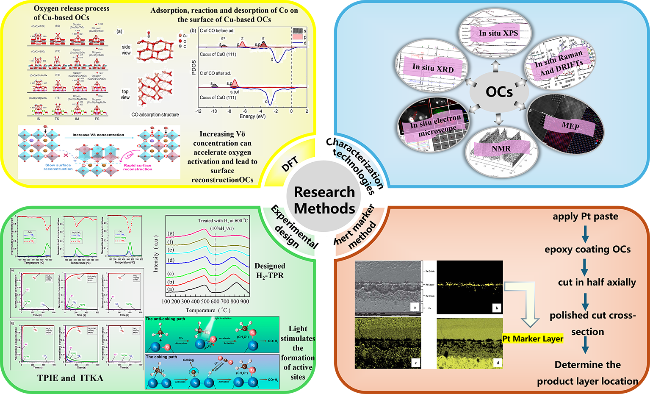
2 The research method to study the mechanism of oxygen storage and release by oxygen carriers
2.1 Advanced characterization Techniques
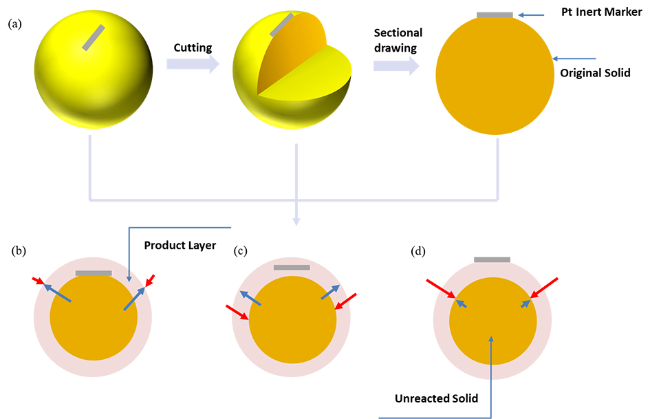
2.2 Experimental design method
2.3 Primary calculation method
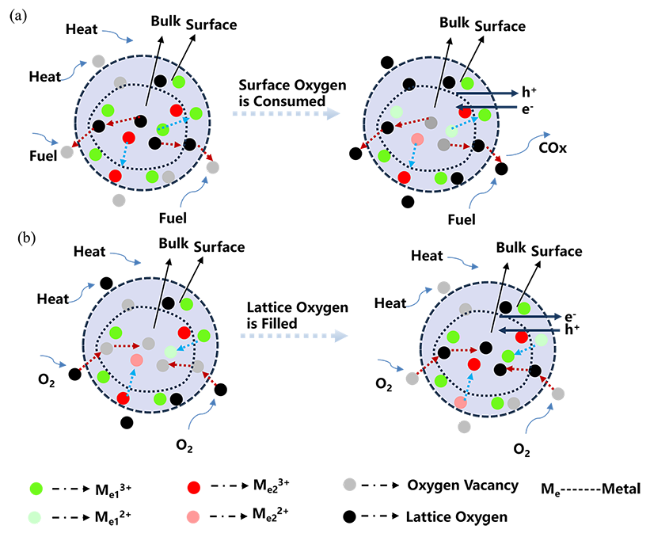
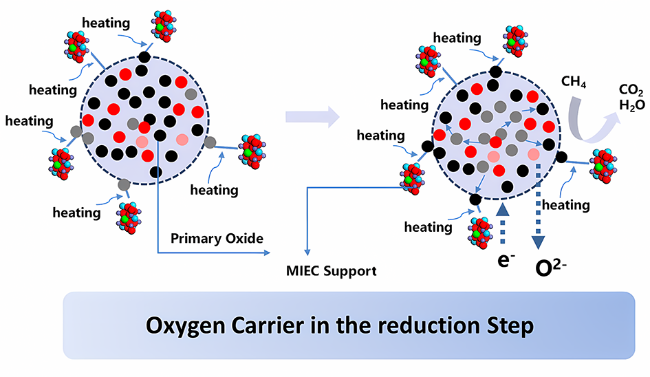
3 Study on lattice oxygen migration mechanism during oxygen storage and release
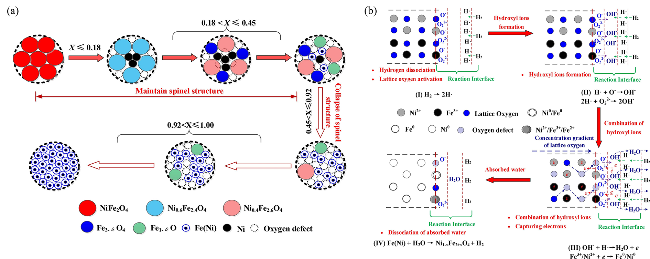
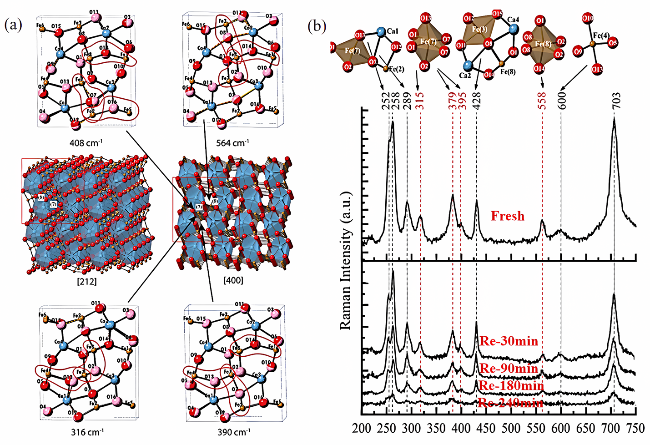
3.1 Lattice oxygen migration mechanism of spinel oxygen carriers
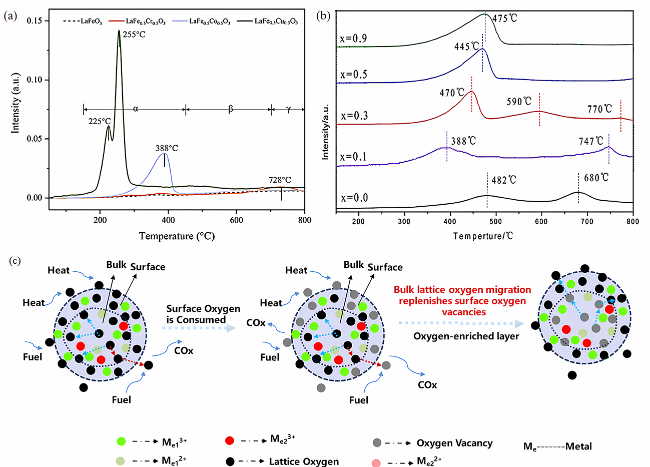
3.2 Lattice oxygen migration mechanism of perovskite-type oxygen carriers
3.3 Lattice oxygen migration mechanism of other metal based oxygen carriers
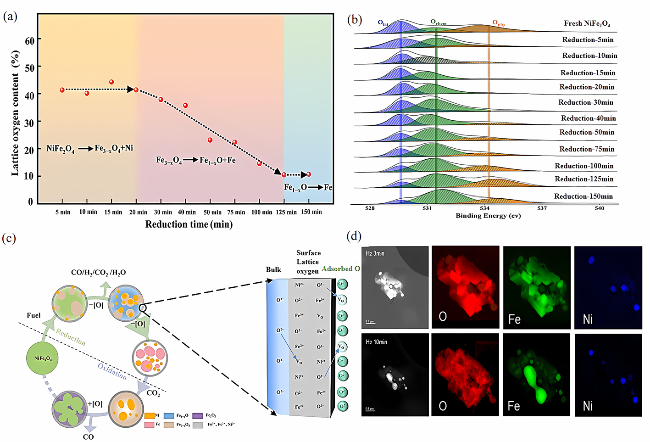
4 Study on metal ions migration mechanism during oxygen storage and release
5 Research limitations in oxygen storage and release processes
5.1 Limitations of the research method
5.2 Limitations of the research mechanism
Nina Chen , Zhiqiang Li , Longyi Guo , Longyu Wen , Lei Jiang , Kongzhai Li . Oxygen Storage and Release Mechanism of Oxygen Carriers[J]. Progress in Chemistry, 2025 , 37(8) : 1156 -1176 . DOI: 10.7536/PC241212
表1 各类表征技术的比较Table 1 Comparison of various characterization techniques |
| Characterization techniques | Advantages | Disadvantages |
|---|---|---|
| In situ XPS[26] | Real-time monitoring of the chemical state on the surface of oxygen carriers and the chemical state in the near surface region provides a strong support for the study of the dynamic evolution of the valence state of the material | The error of quantitative analysis is large and the result is not accurate enough |
| In situ XRD[27] | In situ capture of the crystal evolution of the material | The spatial distribution of polycrystals cannot be obtained on a macroscopic basis |
| In situ Raman[28] | Observe the immediate reaction of substances after receiving external stimuli,and deeply understand the dynamic process inside substances | It is difficult to capture reaction intermediates |
| In situ DRIFTS[29] | Obtain information about the composition,structure and electronic state of the surface without damaging the sample | The signal accuracy is poor at high temperature |
| In situ EPR[30] | It can detect very low concentrations of paramagnetic substances in the sample,with high sensitivity | The temperature must be below 300 ℃,which is not conducive to the high temperature in-situ study of chemical looping |
| Solid high resolution NMR[31] | It can determine the structure,adsorption site and active site of catalyst,and can explore the reaction mechanism through the structure of reactants,intermediates and products | The resolution is poor,the sample carbon spectrum is difficult to peak |
| Atom probe tomography (APT)[32] | It is used to study the phase precipitation process at nanometer scale | Static destructive imaging |
| Synchrotron radiation (XAFS)[33] | It is an experimental technique based on synchrotron radiation light source,which can be used to study the structure,properties and behavior of catalytic materials | There are accuracy problems |
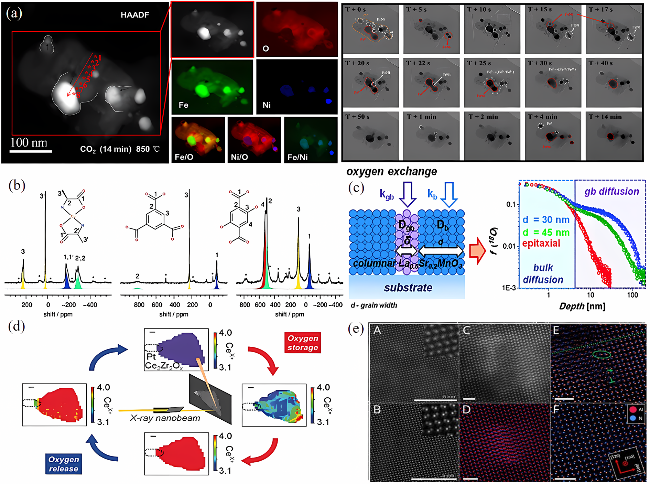
图3 载氧体动态演变先进表征技术:(a) 原位ETEM技术[19];(b) 固体高分辨NMR技术[45]; (c) 18O同位素交换深度剖面分析结合ToF-SIMS技术[46];(d) 原位XAFS成像技术[47];(e) MEP技术[18]Fig. 3 Characterization techniques suitable for the study of migration of active species in the bulk phase of oxygen carriers:(a) in situ ETEM technique[19]; (b) solid-state high-resolution NMR technology[45]; (c) 18O isotope exchange depth profile analysis combined with ToF-SIMS technique[46]; (d) in situ XAFS imaging technique[47]; (e) MEP technique[18] |
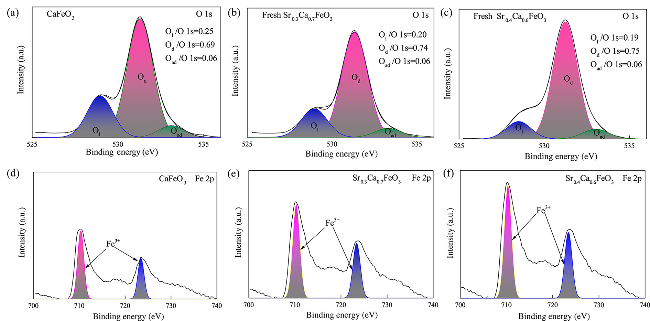
图11 新鲜SrxCa1-xFeO3载氧体的XPS谱图[96]:(a) CaFeO3,O 1s;(b) Sr0.3Ca0.7FeO3,O 1s; (c) Sr0.4Ca0.6FeO3,O 1s; (d) CaFeO3,Fe 2p; (e) Sr0.3Ca0.7FeO3,Fe 2p; (f) Sr0.4Ca0.6FeO3,Fe 2pFig. 11 XPS spectra of fresh SrxCa1-xFeO3[96]:(a) CaFeO3,O 1s;(b) Sr0.3Ca0.7FeO3,O 1s; (c) Sr0.4Ca0.6FeO3,O 1s; (d) CaFeO3,Fe 2p; (e) Sr0.3Ca0.7FeO3,Fe 2p; (f) Sr0.4Ca0.6FeO3,Fe 2p |
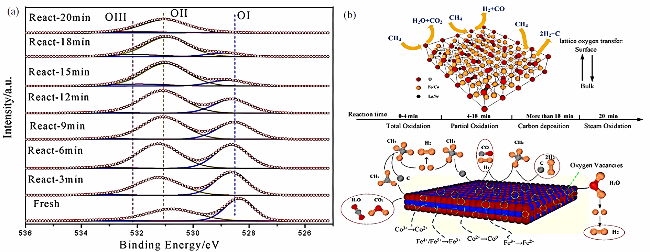
图14 载氧体在甲烷化学链蒸气重整反应中的反应机理[101]:(a) 室温下蒸气氧化的原位XPS谱;(b) 300 ℃蒸气氧化的原位XPS;(c) Fe掺杂的影响机制Fig. 14 Reaction mechanism of oxygen carriers in methane chemical looping steam reforming reaction [101]:(a) in situ XPS spectrum of steam oxidation at room temperature; (b) in situ XPS for steam oxidation at 300 ℃; (c) influence mechanism of Fe doping |
| [1] |
|
| [2] |
|
| [3] |
(段一菲, 陈存壮, 张军社, 王新赫, 魏进家. 中国科学: 化学, 2020, 50(3): 337.).
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
(金红光, 洪慧, 韩涛. 科学通报, 2008, 53(24): 2994.).
|
| [9] |
(王嘉锐, 刘大伟, 邓耀, 徐瑾, 马晓迅, 徐龙. 化工进展, 2024, 43(5): 2235.).
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|
| [24] |
|
| [25] |
|
| [26] |
(赵志娟, 徐鹏, 章小余, 屈宝龙, 刘芬. 中国无机分析化学, 2024, 1.).
|
| [27] |
|
| [28] |
(张艳平, 薛冬峰. 化学研究, 2020, 31(1): 1.).
|
| [29] |
|
| [30] |
|
| [31] |
|
| [32] |
|
| [33] |
|
| [34] |
|
| [35] |
|
| [36] |
|
| [37] |
|
| [38] |
|
| [39] |
|
| [40] |
|
| [41] |
|
| [42] |
(尹钧濂, 冯冉冉, 张月成, 赵继全. 精细化工, 2024, 41(5): 943.).
|
| [43] |
|
| [44] |
|
| [45] |
|
| [46] |
|
| [47] |
|
| [48] |
|
| [49] |
|
| [50] |
|
| [51] |
|
| [52] |
(刘云鹏, 盛伟繁, 吴忠华. 无机材料学报, 2021, 36(9): 901.).
|
| [53] |
(袁妮妮. 宁夏大学博士论文, 2022.).
|
| [54] |
|
| [55] |
(王军民, 杨海波, 武增华, 邱新平, 郭峰. 化学学报, 2003, 61(9): 1410.).
|
| [56] |
|
| [57] |
|
| [58] |
|
| [59] |
|
| [60] |
|
| [61] |
|
| [62] |
|
| [63] |
|
| [64] |
(王志美. 东北大学硕士论文, 2013.).
|
| [65] |
|
| [66] |
|
| [67] |
|
| [68] |
|
| [69] |
|
| [70] |
|
| [71] |
|
| [72] |
|
| [73] |
|
| [74] |
|
| [75] |
|
| [76] |
|
| [77] |
|
| [78] |
|
| [79] |
|
| [80] |
|
| [81] |
|
| [82] |
|
| [83] |
|
| [84] |
|
| [85] |
|
| [86] |
|
| [87] |
|
| [88] |
|
| [89] |
|
| [90] |
|
| [91] |
|
| [92] |
|
| [93] |
|
| [94] |
|
| [95] |
|
| [96] |
|
| [97] |
|
| [98] |
|
| [99] |
|
| [100] |
|
| [101] |
|
| [102] |
|
| [103] |
|
| [104] |
|
| [105] |
|
| [106] |
|
| [107] |
|
| [108] |
|
| [109] |
|
| [110] |
|
| [111] |
|
| [112] |
(李婉莹, 陈良勇. 燃料化学学报, 2024, 52(06): 820.).
|
| [113] |
|
| [114] |
|
| [115] |
|
| [116] |
|
| [117] |
|
| [118] |
|
| [119] |
|
| [120] |
|
| [121] |
|
| [122] |
|
| [123] |
|
| [124] |
|
| [125] |
|
| [126] |
|
| [127] |
|
| [128] |
|
| [129] |
|
| [130] |
|
| [131] |
(张先华. 天津大学博士论文, 2021).
|
/
| 〈 |
|
〉 |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}