
Application of Two-Dimensional Catalysts in Selective Oxidation of Methane
Received date: 2024-12-16
Revised date: 2025-05-07
Online published: 2025-07-25
Supported by
the National Natural Science Foundation of China(22402131)
the National Natural Science Foundation of China(92145301)
the National Natural Science Foundation of China(91845201)
the Fundamental Research Funds for the Liaoning Universities(42400502105)
Doctoral Research Initiation Project of Liaoning Province(2025-BS-0792)
Doctoral Research Initiation Project of Liaoning Province(2025-BS-0970)
Two-dimensional materials,with their high specific surface areas and tunable electronic structures,have shown significant advantages in the enhancement of catalytic efficiency,selectivity,and stability. Their ability to catalyze the conversion of methane into high-value chemicals is of great importance for sustainable energy utilization and environmental protection. This paper reviews the progress of the application of two-dimensional materials in the low-temperature selective oxidation of methane,summarizes the two mechanisms of C—H bond fracture during methane oxidation and lists several typical two-dimensional materials (such as graphene,transition metal sulfides,MXenes,MOFs,metal oxides and their synthesis methods. This paper focuses on investigating the catalytic performance of these materials doped with metal active sites for the selective oxidation of methane using different oxidants (such as H2O2,H2+O2,O2,and CO+O2),emphasizing the role of two-dimensional materials in the regulation of active sites and optimization of reaction pathways. Finally,the potential,challenges and future development direction of two-dimensional materials in solving the problem of methane activation and promoting the progress of energy technology are prospected.
1 Introduction
2 Overview of two-dimensional material catalysts
2.1 Graphene
2.2 Transition metal chalcogenides
2.3 Mxenes
2.4 MOFs
2.5 Metal oxides
2.6 Other materials
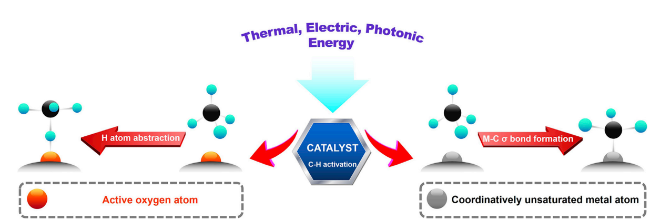
3 Mechanism of C—H bond cleavage of methane oxidation
3.1 Radical mechanism
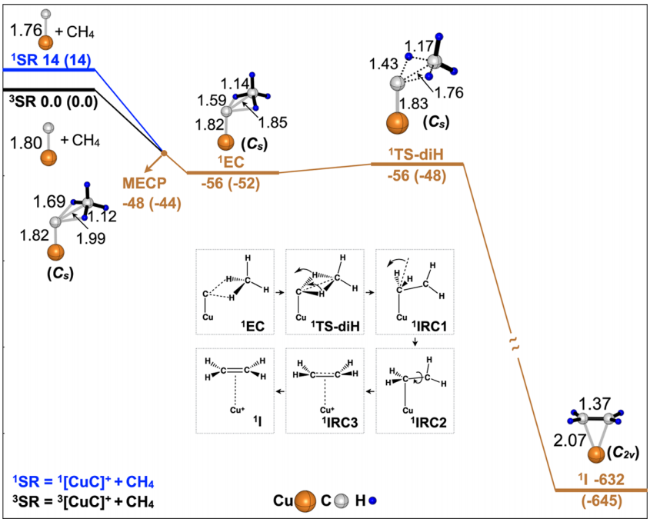
3.2 M-C σ-bond mechanism
4 The application of two-dimensional materials doped metal atom catalysts in methane oxidation reaction
4.1 Precious metals
4.2 Non-precious metals
4.3 Two-dimensional materials doped with metal atom catalysts
5 Conclusion and outlook
Yuyang Sun , Wenxi Wang , Wencui Li , Hanying Qin , Jiaxin Cai , Zhen Zhao . Application of Two-Dimensional Catalysts in Selective Oxidation of Methane[J]. Progress in Chemistry, 2025 , 37(8) : 1218 -1234 . DOI: 10.7536/PC241206
表1 不同类型催化剂温和条件下选择性氧化甲烷性能比较Table 1 Comparison of selective methane oxidation performance under mild conditions of different types of catalysts |
| No. | Catalysts | Reaction conditions | Selectivity (%) | Oxygenated product yield (mmol·g-1·h-1) | Ref. |
|---|---|---|---|---|---|
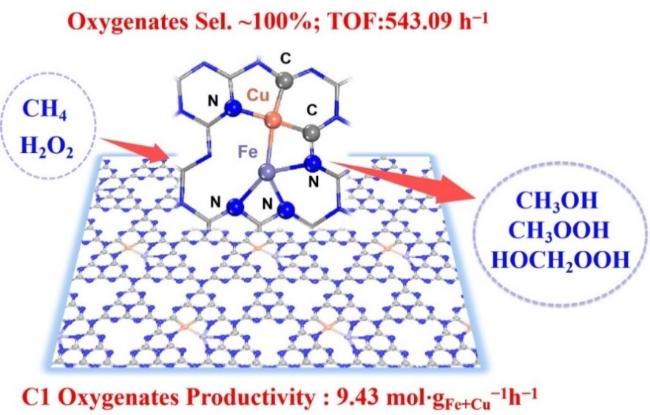
| 1 | Fe1/Cu1-C3Nx | Cat:3 mg; OX:H2O2; T:80 ℃; P:3 MPa CH4; t:30 min | ≈100 | 9430 | 77 |
| 2 | Ru1/UiO-66 | Cat:0.13 wt%+0.72 wt%; OX:H2O2; T:60 ℃; P:3 MPa CH4; t:24 h | ≈100 | 3.7027 | 94 |
| 3 | Fe/MIL-53(Al) | Cat:0.3~5.5 wt%; OX:H2O2; T:60 ℃; P:/; t:1 h | 80 | / | 95 |
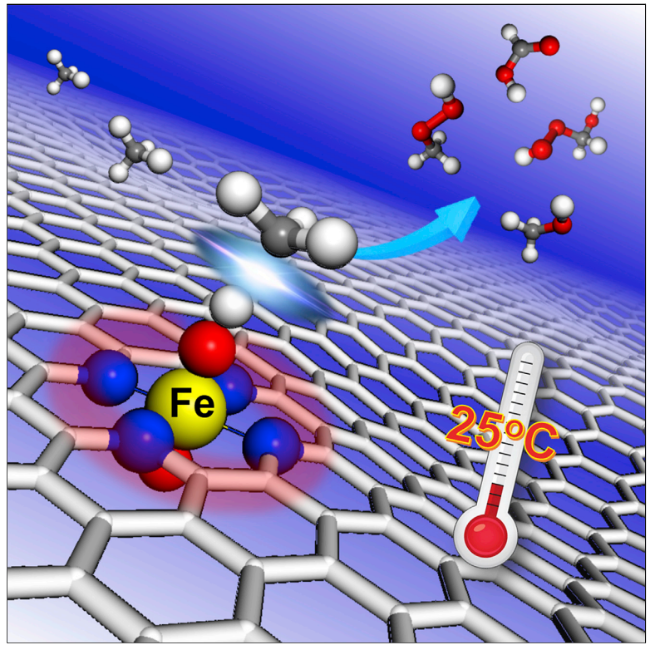
| 4 | FeN4/GN | Cat:50 mg; OX:H2O2; T:25 ℃; P:2 MPa CH4; t:10 h | 94 | / | 118 |
| 5 | FeOx/TiO2 | Cat:10 mg; OX:H2O2; T:25 ℃; P:1 bar CH4+Ar; t:3 h | 97 | 1056 | 91 |
| 6 | SACs Rh-CeO2 NWS | Cat:10 mg; OX:H2O2; T:50℃; P:0.5 MPa CH4; t:1 h | 93.9 | 1231.7 | 121 |
| 7 | Cu-BTC-P-235 | Cat:25 mg; OX:H2O2; T: 50 ℃; P: 3 MPa CH4; t:10 min | 99.6 | 10.67 | 93 |
| 8 | CUS-M-P-210 | Cat:10 mg; OX:H2O2; T:80℃; P:3 MPa CH4; t:1 h | 100 | 83.13 | 122 |
| 9 | Fe-ZSM-5 | Cat:50 mg; OX:H2+O2; T:30 ℃; P:15 bar CH4+3 bar H2+10 bar O2; t:4 h | 94 | / | 96 |
| 10 | Pd-Au/CNTs | Cat:30 mg; OX:H2+O2; T:50 ℃; P:3. 3MPa CH4; t:30 min | 73.2 | 190.1 | 126 |
| 11 | PtOx/BN-na | Cat:0.02 g; OX:O2; T:150 ℃; P:O2+CH4+H2O; t:2.5 h | 95 | 106.5 | 86 |
| 12 | Au/ZnO | Cat:10 mg; OX:O2; T:25 ℃; P:2 MPa CH4+0.1 MPa O2; t:2 h | 95 | 0.125 | 97 |
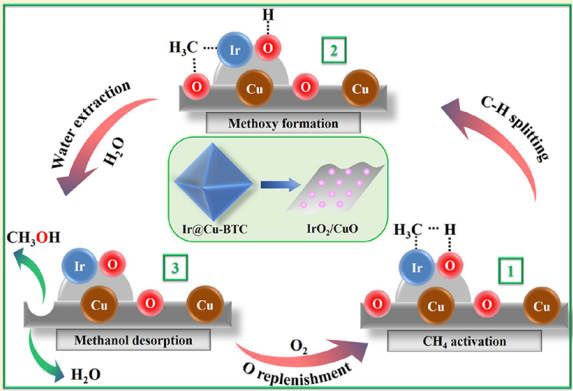
| 13 | IrO2/CuO | Cat:0.01 g; OX:O2; T:150 ℃; P:20 bar CH4; t:3 h | 95 | 1.937 | 130 |
| 14 | Au1/BP | Cat:0.2 wt%; OX:O2; T:90 ℃; P:30 bar CH4+3 bar O2; t:2 h | >99 | 0.1135 | 131 |
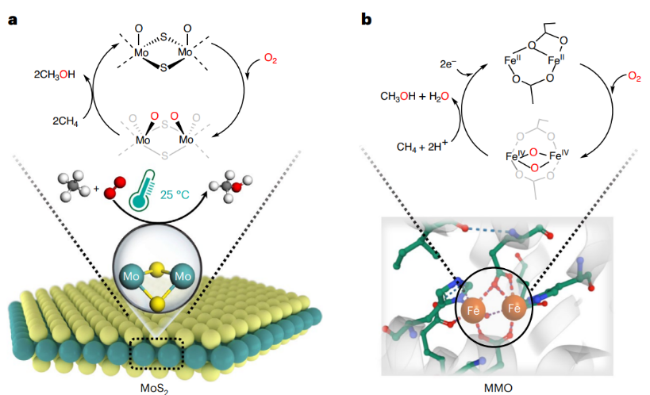
| 15 | ER-MoS2 | Cat:200 mg; OX:O2; T:25 ℃; P:5 bar CH4+1 bar O2; t:24h | >99 | 0.0407 | 132 |
| 16 | Au/ZSM-5 | Cat:0.1 g; OX:O2; T:120~240 ℃; P:20. 7 bar CH4+3.5 bar O2; t:4 h | ≈100 | 0.545 | 98 |
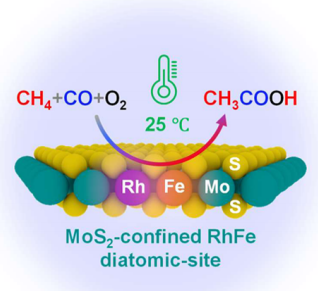
| 17 | Rh/ZSM-5 | Cat:20 mg; OX:O2+CO; T:150 ℃; P:30 bar>CH4+O2+CO; t:3 h | 60~100 | 22.23 | 99 |
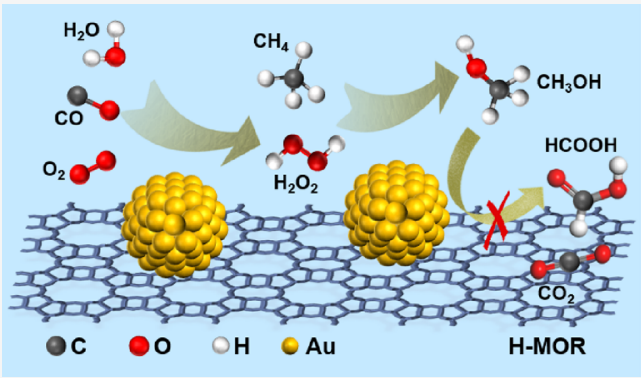
| 18 | Au/H-MOR | Cat:0.1 g; OX:O2+CO; T:150 ℃; P:30 bar CO+O2+CH4; t:1 h | 75 | 280 | 137 |
“/”文献未提供数据. |
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|
| [24] |
|
| [25] |
|
| [26] |
|
| [27] |
(王磊, 于新海, 袁帅帅, 姚馨淇, 李传东. 材料导报, 2023, 37(21): 94.).
|
| [28] |
|
| [29] |
|
| [30] |
|
| [31] |
|
| [32] |
|
| [33] |
|
| [34] |
|
| [35] |
|
| [36] |
|
| [37] |
|
| [38] |
|
| [39] |
(陈芸, 张辉, 罗政, 毛卫国, 潘俊安, 王珊珊. 化学进展, 2024, 36(04): 537.).
|
| [40] |
|
| [41] |
(刘慧敏, 李克智, 张欣, 殷学民, 付前刚, 李贺军. 物理化学学报, 2024, 40(02): 64.).
|
| [42] |
(吴正颖, 刘谢, 刘劲松, 刘守清, 查振龙, 陈志刚. 化学进展, 2019, 31(08): 1086.).
|
| [43] |
|
| [44] |
|
| [45] |
|
| [46] |
|
| [47] |
(李煜斌, 代国亮, 范杰, 肖红. 化学进展, 2024, 36(09): 1336.).
|
| [48] |
|
| [49] |
|
| [50] |
|
| [51] |
|
| [52] |
|
| [53] |
|
| [54] |
|
| [55] |
|
| [56] |
|
| [57] |
|
| [58] |
|
| [59] |
|
| [60] |
|
| [61] |
|
| [62] |
|
| [63] |
|
| [64] |
|
| [65] |
|
| [66] |
|
| [67] |
|
| [68] |
|
| [69] |
|
| [70] |
|
| [71] |
(吴怡枭, 刘超波, 昝雪玉, 张超宇, 陶诗琪, 李智雯, 王柯静, 刘勇军, 黄伟. 高等学校化学学报, 2024, 45(09): 9.).
|
| [72] |
(张金水, 王博, 王心晨. 物理化学学报, 2013, 29(9): 1865.)
|
| [73] |
|
| [74] |
|
| [75] |
(查向浩, 安旭霞, 李飞星, 李有文, 张玉才. 化工新型材料, 2024, 52(09): 31.).
|
| [76] |
|
| [77] |
|
| [78] |
|
| [79] |
|
| [80] |
|
| [81] |
|
| [82] |
|
| [83] |
|
| [84] |
(罗曼, 周扬, 成田恬, 孟雨欣, 王奕锦, 鲜佳赤, 秦嘉怡, 余晨辉. 光子学报, 2024, 53(7): 0753307.).
|
| [85] |
(程世群, 翁雪霏, 王庆楠, 周百川, 李文翠, 李名润, 贺雷, 王东琪, 陆安慧. 催化学报, 2022, 43(4): 1092.).
|
| [86] |
|
| [87] |
|
| [88] |
|
| [89] |
|
| [90] |
|
| [91] |
|
| [92] |
|
| [93] |
|
| [94] |
|
| [95] |
|
| [96] |
|
| [97] |
|
| [98] |
|
| [99] |
|
| [100] |
|
| [101] |
|
| [102] |
|
| [103] |
|
| [104] |
|
| [105] |
|
| [106] |
(申恒芳, 李文翠, 蔡佳芯, 秦汉颖, 张航, 赵震. 中国科学: 化学, 2024, 54(3): 309.).
|
| [107] |
(马立超, 王俊文, 徐红. 低碳化学与化工, 2023, 48(01): 58.).
|
| [108] |
(何泽召, 杨克武, 蔚翠, 李佳, 刘庆彬, 芦伟立, 冯志红, 蔡树军. 中国物理快报, 2015, 32(11): 122.).
|
| [109] |
|
| [110] |
|
| [111] |
|
| [112] |
|
| [113] |
|
| [114] |
|
| [115] |
|
| [116] |
|
| [117] |
|
| [118] |
|
| [119] |
|
| [120] |
|
| [121] |
|
| [122] |
|
| [123] |
|
| [124] |
|
| [125] |
|
| [126] |
|
| [127] |
|
| [128] |
|
| [129] |
|
| [130] |
|
| [131] |
|
| [132] |
|
| [133] |
|
| [134] |
|
| [135] |
|
| [136] |
|
| [137] |
|
| [138] |
|
| [139] |
|
/
| 〈 |
|
〉 |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}