
Development of Mirror-Image Protein Drugs: Advances in Chemical Synthesis, Mirror-Image Phage Display, and Computational Design
Received date: 2025-03-26
Revised date: 2025-04-29
Online published: 2025-09-01
Supported by
The National Natural Science Foundation of China(22307061)
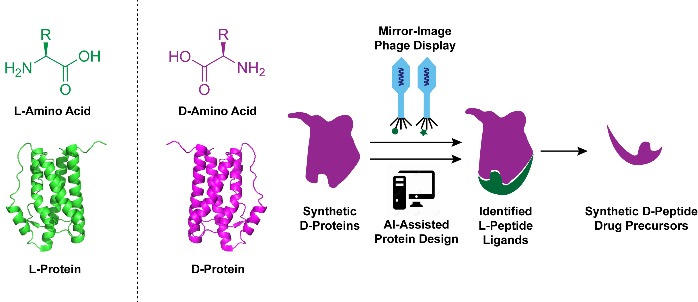
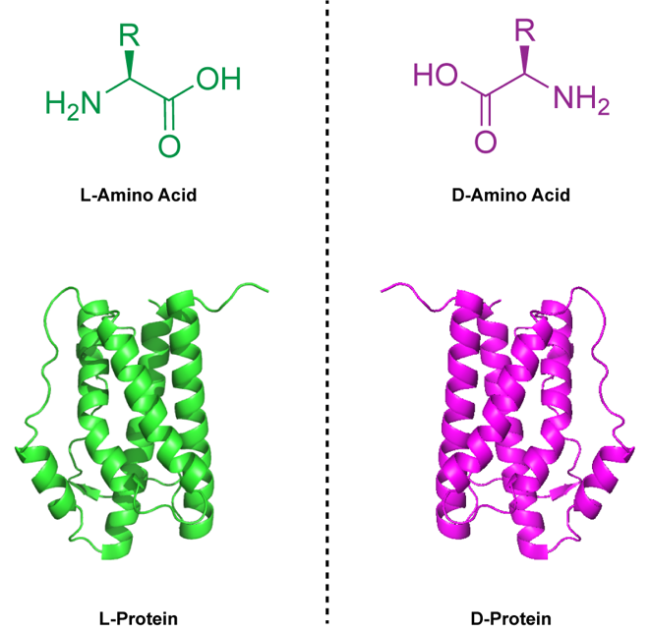
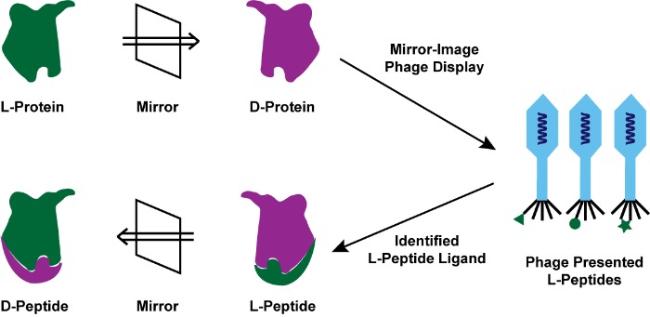
Mirror-image peptides and proteins composed entirely of D-amino acids have emerged as promising therapeutic candidates owing to their resistance to proteolysis and reduced immunogenicity. Mirror-image phage display (MIPD) is currently the main experimental technique for identifying mirror-image peptide ligands targeting disease-related proteins. However, the success of MIPD critically depends on synthetic mirror-image target proteins, which cannot be produced by traditional recombinant methods due to the intrinsic chirality of biological systems. Recent advances in chemical protein synthesis, such as enzyme-cleavable solubilizing tags, backbone-installed split intein-assisted ligation, and removable glycosylation modification-assisted folding strategies, have effectively addressed key challenges in preparing these complex mirror-image proteins. In addition, computational approaches, exemplified by AI-driven protein design, have become powerful complementary tools, accelerating the discovery and optimization of mirror-image protein drug candidates. Although mirror-image protein drugs have not yet reached clinical use, ongoing innovations in chemical synthesis and ligand screening methods are steadily advancing their therapeutic potential toward clinical translation.
1 Introduction
2 Mirror-image phage display
3 Chemical protein synthesis
3.1 Solid-phase peptide synthesis
3.2 Native chemical ligation
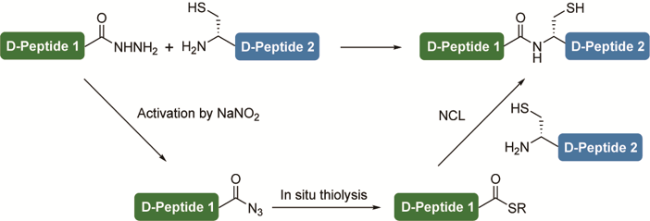
3.3 Peptide hydrazide ligation
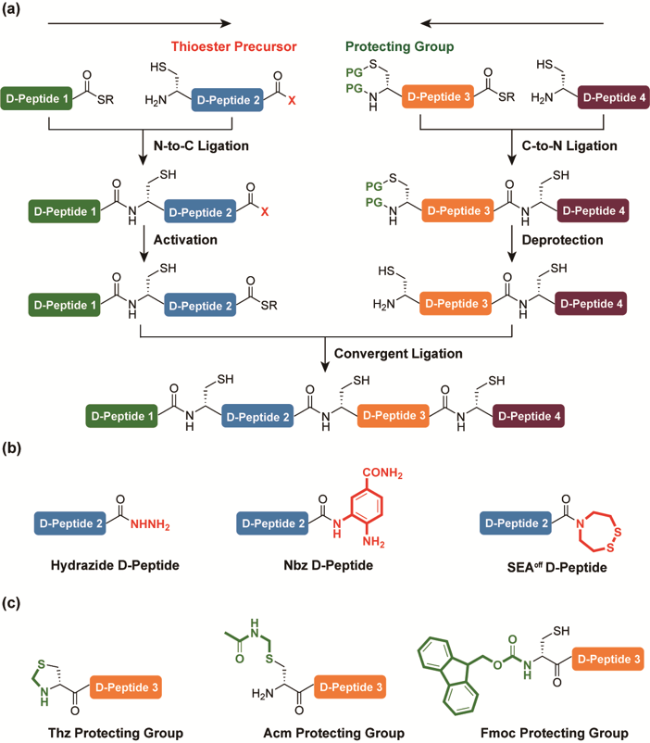
3.4 Multiple-segment ligation
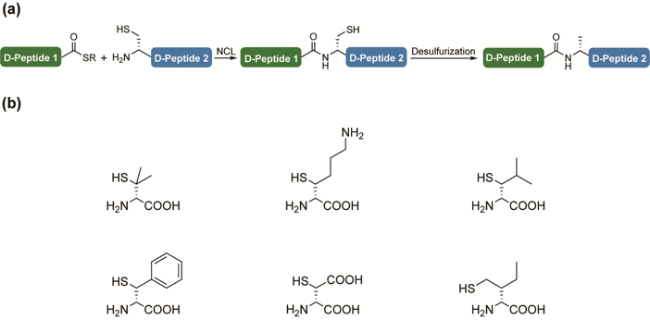
3.5 The ligation-desulfurization strategy
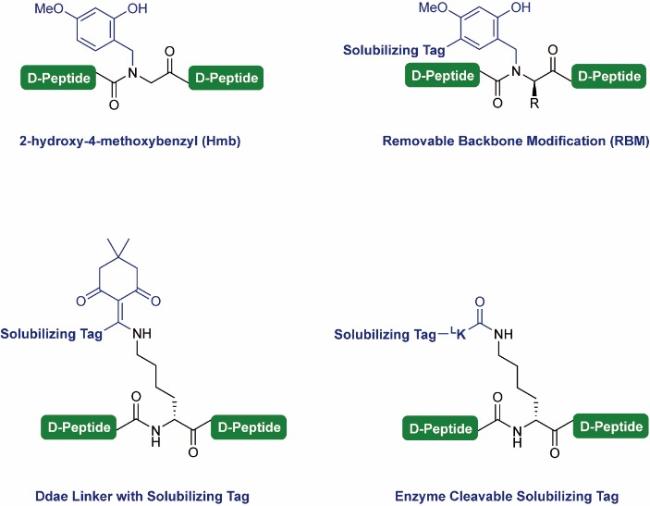
3.6 Solubilizing tags for hydrophobic segment
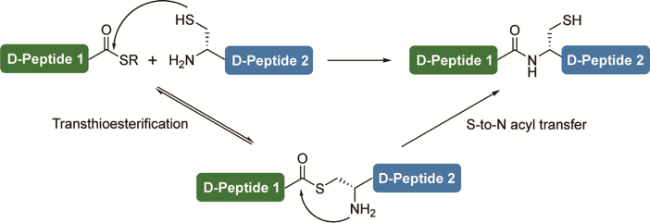
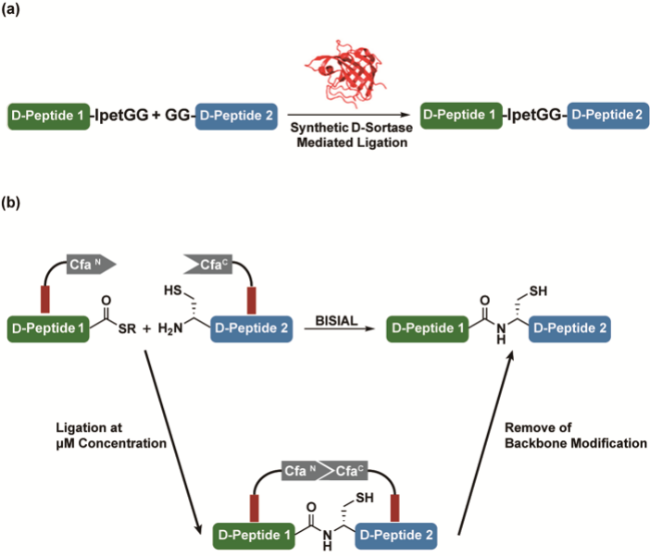
3.7 Chemoenzymatic D-peptide ligation
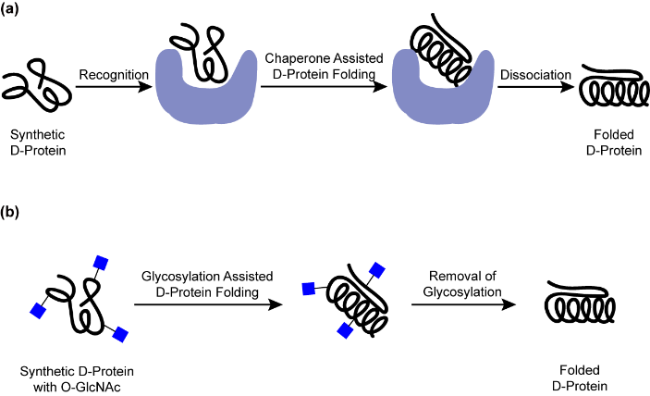
3.8 The folding of D-protein
4 Applications of mirror-image phage display
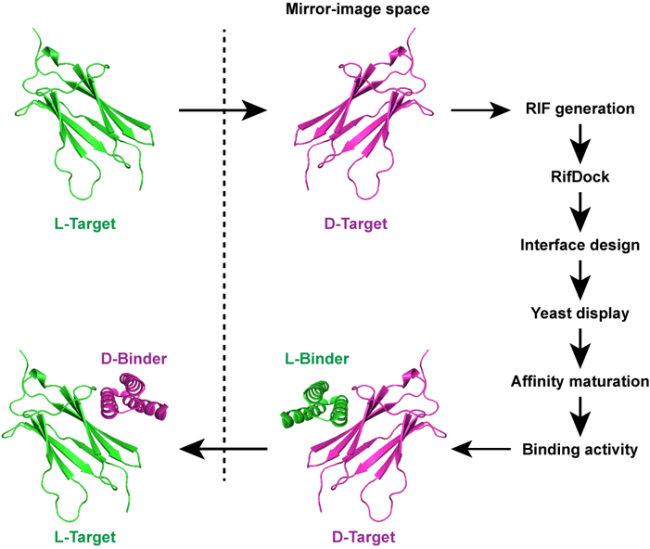
5 Computationally assisted discovery of mirror-image protein drugs
6 Conclusion and outlook
Ren Yuxiang , Han Dongyang , Shi Weiwei . Development of Mirror-Image Protein Drugs: Advances in Chemical Synthesis, Mirror-Image Phage Display, and Computational Design[J]. Progress in Chemistry, 2025 , 37(9) : 1261 -1273 . DOI: 10.7536/PC20250315
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|
| [24] |
|
| [25] |
|
| [26] |
|
| [27] |
|
| [28] |
|
| [29] |
|
| [30] |
|
| [31] |
|
| [32] |
|
| [33] |
|
| [34] |
|
| [35] |
|
| [36] |
|
| [37] |
|
| [38] |
|
| [39] |
|
| [40] |
|
| [41] |
|
| [42] |
|
| [43] |
|
| [44] |
|
| [45] |
|
| [46] |
|
| [47] |
|
| [48] |
|
| [49] |
|
| [50] |
|
| [51] |
|
| [52] |
|
| [53] |
|
| [54] |
|
| [55] |
|
| [56] |
|
| [57] |
|
| [58] |
|
| [59] |
|
| [60] |
|
| [61] |
|
| [62] |
|
| [63] |
|
| [64] |
|
| [65] |
|
| [66] |
|
| [67] |
|
| [68] |
|
| [69] |
|
| [70] |
|
| [71] |
|
| [72] |
|
| [73] |
|
| [74] |
|
| [75] |
|
| [76] |
|
| [77] |
|
| [78] |
|
| [79] |
|
| [80] |
|
| [81] |
|
| [82] |
|
| [83] |
|
| [84] |
|
| [85] |
|
| [86] |
|
| [87] |
|
| [88] |
|
| [89] |
|
| [90] |
|
| [91] |
|
| [92] |
|
| [93] |
|
| [94] |
|
| [95] |
|
| [96] |
|
| [97] |
|
| [98] |
|
| [99] |
|
| [100] |
|
| [101] |
|
| [102] |
|
| [103] |
|
| [104] |
|
| [105] |
|
| [106] |
|
| [107] |
|
| [108] |
|
| [109] |
|
| [110] |
|
| [111] |
|
| [112] |
|
| [113] |
|
| [114] |
|
| [115] |
|
| [116] |
|
| [117] |
|
| [118] |
|
| [119] |
|
| [120] |
|
| [121] |
|
| [122] |
|
| [123] |
|
| [124] |
|
| [125] |
|
| [126] |
|
| [127] |
|
| [128] |
|
| [129] |
|
| [130] |
|
| [131] |
|
| [132] |
|
| [133] |
|
| [134] |
|
| [135] |
|
| [136] |
|
| [137] |
|
| [138] |
|
| [139] |
|
| [140] |
|
| [141] |
|
| [142] |
|
| [143] |
|
| [144] |
|
| [145] |
|
| [146] |
|
| [147] |
|
| [148] |
|
| [149] |
|
| [150] |
|
| [151] |
|
| [152] |
|
| [153] |
|
| [154] |
|
| [155] |
|
| [156] |
|
| [157] |
|
| [158] |
|
| [159] |
|
| [160] |
|
| [161] |
|
| [162] |
|
| [163] |
|
/
| 〈 |
|
〉 |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}