
Gas Phase Selective Oxidation of Methane to Formaldehyde
Received date: 2025-02-05
Revised date: 2025-04-25
Online published: 2025-09-01
Supported by
The National Natural Science Foundation of China(22172101)
The Applied Basic Research Program of Liaoning Province(2023JH2/101600059)
The "Xingliao Talents" Youth Top Talent Program of Liaoning Province(XLYC2203138)
The Special Fund for Basic Scientific Research and Operation of undergraduate universities in Liaoning Province(LJ212410166046)
The Major Project Incubation Project of Shenyang Normal University
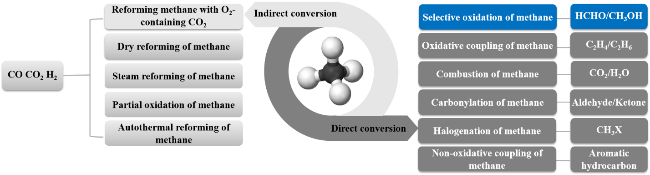
Methane, as a light alkane clean resource with abundant reserves, its efficient utilization has significant practical significance. Direct conversion of methane into high-value target products through gas-phase selective oxidation of methane has become an effective way to efficiently utilize methane. This reaction has the advantages of simple equipment and relatively low reaction energy consumption. However, the strong carbon-hydrogen bond of methane makes its activation process difficult, and the product formaldehyde is prone to deep oxidation under high-temperature and oxygen-containing conditions, resulting in a decrease in the selectivity of the target product. Therefore, achieving high-selectivity direct oxidation of methane to form oxygen-containing compounds is challenging. This article reviews the research progress in the gas-phase selective oxidation of methane to formaldehyde, focusing on the reaction mechanism of selective oxidation of methane to formaldehyde on catalysts, catalyst systems, and the application of various in-situ characterizations in the reaction. Finally, the future development directions of the selective oxidation of methane are summarized and prospected.
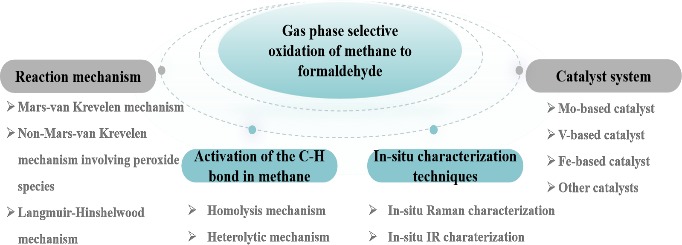
1 Introduction
2 Methane C―H bond activation
3 Reaction mechanism of gas phase selective oxidation of methane to formaldehyde
3.1 Mars-van Krevelen mechanism
3.2 Non‑Mars‑van Krevelen mechanism involving peroxide species
3.3 Langmuir‑Hinshelwood mechanism
4 Methane selective oxidation reaction catalyst system
4.1 Mo‑based catalyst
4.2 V‑based catalyst
4.3 Fe‑based catalyst
4.4 Other catalysts
5 In‑situ characterization of methane selective oxidation reaction
6 Conclusion and outlook
Key words: methane; gas-phase selective oxidation; formaldehyde; reaction mechanism; catalyst
He Yan , Song Jiaxin , Fan Xiaoqiang , Yu Xuehua , Zhao Zhen . Gas Phase Selective Oxidation of Methane to Formaldehyde[J]. Progress in Chemistry, 2025 , 37(9) : 1321 -1341 . DOI: 10.7536/PC20250201
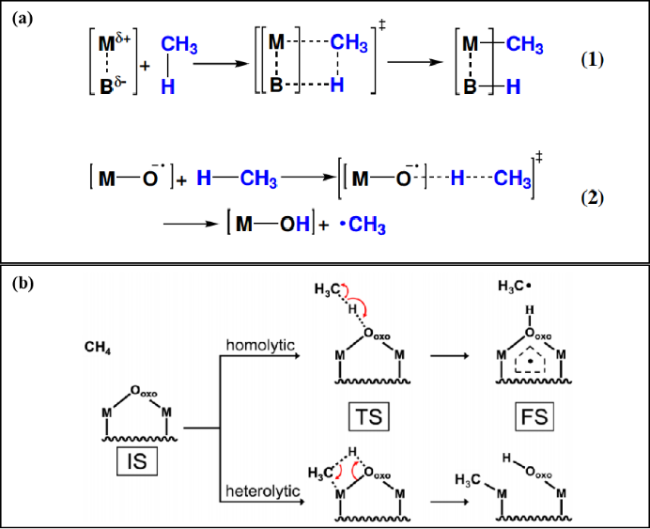
图2 (a) 碳氢键断裂的潜在途径: (1) 异裂; (2) 均裂[41]; (b) 金属氧化物纳米团簇上甲烷活化的均裂和异裂机制的初始态(IS), 过渡态(TS)和最终态(FS)[44]Fig.2 (a) Potential mechanistic pathways for C―H bond cleavage. (1) Heterolytic; (2) homolytic[41]; (b) initial state (IS), transition state (TS), and final state (FS) of homolytic and heterolytic mechanisms for methane activation over a metal oxide nanocluster[44] |
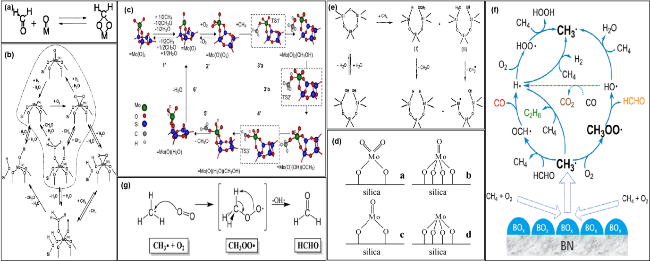
图3 (a) 二氧亚甲基(表面缩醛)中间体的可逆形成[56]; (b) 甲烷在孤立的SiO2担载的MoOx催化剂上的氧化机制, 包括由H2O存在的平行路径[61]; (c) 基于=Mo(O)2为活性中心的反应机理. 虚线框表示过渡态结构[62]; (d) 二氧化硅上孤立的钼(Ⅵ) (a, b)和钼(Ⅳ) (c, d)氧化物物种结构[65];(e) 二氧化硅表面的脱羟基化和二氧化硅催化剂上甲烷在硅氧烷缺陷位点活化生成气相甲醛和甲基自由基的反应途径[72]; (f) h-BN催化剂表面自由基引发甲烷氧化的气相反应网络[73]; (g) 气相中CH3·转化为CH3OO·和HCHO的可能途径[73]Fig.3 (a) Reversible formation of dioxymethylene (surface acetal) intermediates[56]; (b) proposed mechanism of CH4 oxidation at isolated, SiO2-supported MoOx sites, including parallel pathways enabled by the presence of H2O[61]; (c) proposed reaction mechanism based on the assumption of =Mo(O)2 as the active center. Transition state structures are enclosed within dotted lines[62]; (d) proposed structures for the isolated Mo(VI) (a, b) and Mo(IV) (c, d) oxide species on silica[65]; (e) dehydroxylation of the silica surface and the reaction pathway for methane activation at the siloxane defect site over silica catalysts to yield gas phase formaldehyde and methyl radicals[72]; (f) proposed Gas-Phase Reaction Network for Radical-Triggered Methane Oxidation over the h-BN Catalyst Surface[73]; (g) proposed Pathway of CH3· Conversion to CH3OO· and HCHO in the Gas Phase[73] |
表1 甲烷选择氧化反应催化剂的催化活性Table 1 Catalytic activity of catalysts for selective oxidation of methane |
| Catalyst | Reaction condition | CH4 Conversion | HCHO Selectivity | HCHO Yield | Ref | ||
|---|---|---|---|---|---|---|---|
| K2MoO4/SiO2 | 650 ℃ | GHSV=6000 h-1 | CH4/O2=9 | 1.3% | 32.1% | 0.42% | 70 |
| 1%V2O5/3%MoO3/SiO2 | 630 ℃ | GHSV=70 000 L·kg-1·h-1 | CH4/air=1.5 | 8.47mol% | 16.6mol% | - | 77 |
| MoO3-AWSG (acid-washed silica gel) | 650 ℃ | GHSV=5000 h-1 | CH4/O2=9 | 6.9% | 76% | 0.04% | 80 |
| 0.2wt%MoO3/SiO2 | 520 ℃ | - | CH4/O2=2 | - | 41% | - | 82 |
| 9.9 wt%Mo-SBA-1 | 640 ℃ | - | CH4/O2=4.5 | 8.2% | 20% | 1.64% | 85 |
| Mo/HZSM-5 | 600 ℃ | GHSV=7.41 g·h·mol-1 | CH4/O2=5.3 | 13.1mol% | 3.4mol% | - | 86 |
| 8%Mo/MCM-22 | 650 ℃ | GHSV=18 000 L·kg-1·h-1 | CH4/O2=2 | 3.8% | 12.2% | 0.46% | 87 |
| 8Mo-KIT-6 | 675 ℃ | GHSV=36 000 mL·g·h-1 | CH4/O2=2 | 7.3% | 28.8% | 2.1% | 88 |
| 3 wt%VOx/SBA-15 | 625 ℃ | GHSV=72 000 mL·g·h-1 | - | 4.6% | 81% | 3.7% | 92 |
| 12 wt%Mo/ZrO2 | 400 ℃ | GHSV=12 000 mL·g-1·h-1 | CH4/O2=10 | 8.3% | 47.8% | 4% | 100 |
| Cu-MoOx | 650 ℃ | GHSV=84 000 mL·g·h-1 | - | 0.3% | 77% | 0.23% | 103 |
| VOx/MCM-41 | 868 ℃ | GHSV=180 000 L·kg-1·h-1 | CH4/air=1.14 | 3.2% | 29.1% | 0.93% | 107 |
| 1%V2O5/SiO2 | 630 ℃ | GHSV=70 000 L·kg-1·h-1 | CH4/air=1.5 | 9.52% | 15.7% | 1.49% | 110 |
| V/SBA-15 | 600 ℃ | GHSV=35 300 L·kg-1·h-1 | CH4/O2=2 | 15.2% | 15.8% | 2.41% | 120 |
| V/Ti-SBA-15 | - | GHSV=360 000 mL·g·h-1 | CH4/O2=9 | 0.25% | 75% | 0.18% | 126 |
| 3V-KIT-6 | 625 ℃ | - | CH4/O2=2 | 8.1% | 26.1% | 2.11% | 175 |
| 4.2V-DMSN | 530 ℃ | - | - | 0.6% | 76.3% | 0.46% | 179 |
| Fe-MCM-41 | 500 ℃ | - | - | 2% | 13.6% | 0.27% | 124 |
| P-FeOx-SiO2(P/Fe=0.5) | 625 ℃ | - | - | 6% | 40% | 2.4% | 134 |
| FeOx/SBA-15 | 650 ℃ | GHSV=72 000 mL·g·h-1 | - | 5% | 37% | 1.9% | 57 |
| FeOx/SiO2 | 650 ℃ | GHSV=60 000 mL·g·h-1 | - | 37% | 33% | 12.2% | 137 |
| 20 wt%FePO4/MCM-41 | 500 ℃ | GHSV=21 000 mL·g·h-1 | - | 0.41% | 79.2% | 0.32% | 142 |
| FePO4 | 500 ℃ | GHSV=36 000 mL·g·h-1 | - | 0.51% | 39% | 0.2% | 146 |
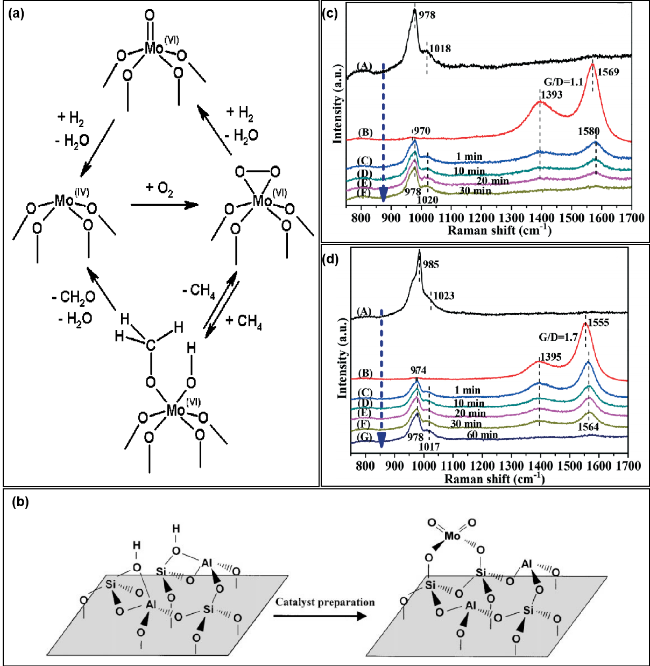
图4 (a) 甲烷在二氧化硅负载孤立的MoOx位点上的氧化机理[61]; (b) 四面体单体Mo物种与沸石Brönsted酸位的连接模型[86];脱水后的8Mo-KIT-6 (c)和4.6Mo/KIT-6 (d)催化剂在675 ℃, (A) 16.7%O2/N2的氧化流量和(B) 33.3%CH4/N2的还原流量的条件下, 在(C) 1 min、(D) 10 min、(E) 20 min、(F) 30 min、(G) 60 min的真实反应条件下进行的原位紫外拉曼光谱研究[88]Fig. 4 (a) Proposed mechanism of CH4 oxidation at isolated, SiO2-supported MoOx sites[61]; (b) model of the joining of the tetrahedral monomeric Mo species to the Brönsted acid sites of the zeolite[86]; in situ UV Raman spectra of the dehydrated 8Mo-KIT-6 (c) and 4.6Mo/KIT-6 (d) catalysts at 675 ℃ under (A) oxidizing flow of 16.7% O2/N2, and under (B) reducing flow of 33.3% CH4/N2, and exposed to real reaction condition of 33.3% CH4/16.7% O2/N2 after (C) 1 min, (D) 10 min, (E) 20 min, (F) 30 min, (G) 60 min[88] |
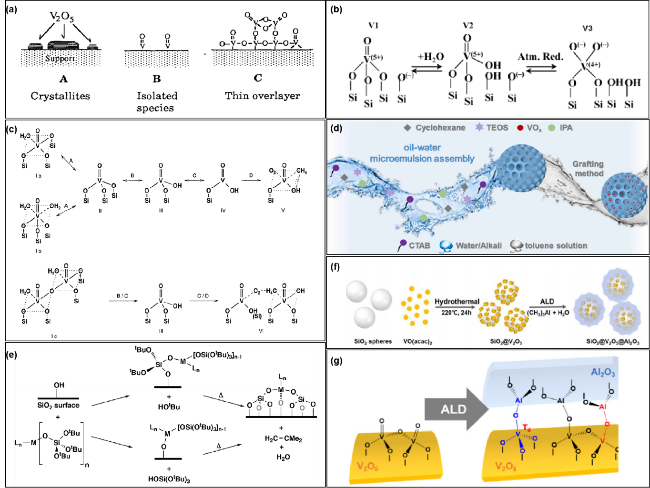
图5 (a) 载体上VOx覆盖层种类[109]; (b) 非羟基化(V1)和羟基化(V2)物种之间以及羟基化(V2)物种和活性位点(V3)之间的平衡示意图[111]; (c) VOx/MCM催化剂活性位点的形成过程及其与O2和CH4的相互作用[112]; (d) V/MSN催化剂形成机理示意图[113]; (e) 通过热裂解分子前驱体方法获得的表面负载孤立活性位点[125]; (f) SiO2@V2O5@Al2O3核壳纳米结构制备示意图[127]; (g) V2O5和Al2O3在SiO2@V2O5@Al2O3催化剂中发生反应所形成新的Td钒物种和V―O―Al键[127]Fig.5 (a) Models of vanadium oxide overlayers on supports[109]; (b) schematic equilibriums between non-hydroxylated (V1) and hydroxylated (V2) species and between hydroxylated (V2) species and proposed active sites (V3)[111]; (c) scheme of the formation of the active sites and their interaction with O2 and CH4[112]; (d) the schematic diagram for the formation mechanism of V/MSN catalysts[113]; (e) thermolytic molecular precursor approach to surface-supported, isolated active sites[125]; (f) schematic preparation of SiO2@V2O5@Al2O3 core@shell nanostructures[127]; (g) formation of new Td vanadium species and V―O―Al bonds by a reaction between V2O5 and Al2O3 in the SiO2@V2O5@Al2O3 core@shell catalyst[127] |
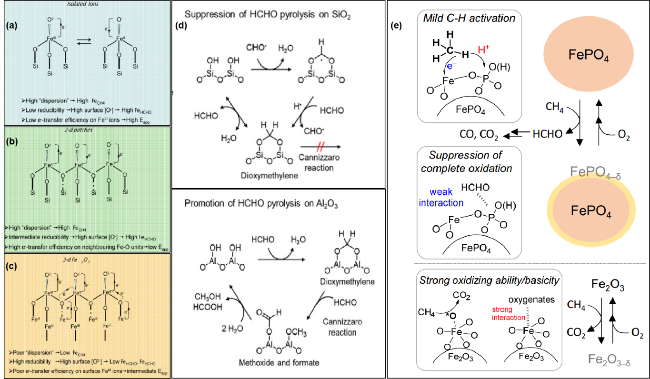
图6 (a~c) FeOx/SiO2催化剂的表面结构和对应反应模式[137]; (d) 提出了抑制甲醛在SiO2上热解和促进甲醛在Al2O3上热解的机理[143]; (e) 提出了FePO4上的甲烷部分氧化成甲醛和Fe2O3上的甲烷完全氧化成CO2的反应机理[146]Fig.6 (a~c) Surface structures of FeOx/SiO2 catalysts and relative reactivity pattern[137]; (d) proposed mechanism of the suppression of HCHO pyrolysis on SiO2 and the promotion of HCHO pyrolysis on Al2O3[143]; (e) proposed reaction mechanism for oxidation of CH4 over FePO4 into HCHO with O2 and complete oxidation of CH4 over Fe2O3 into CO2 with O2[146] |
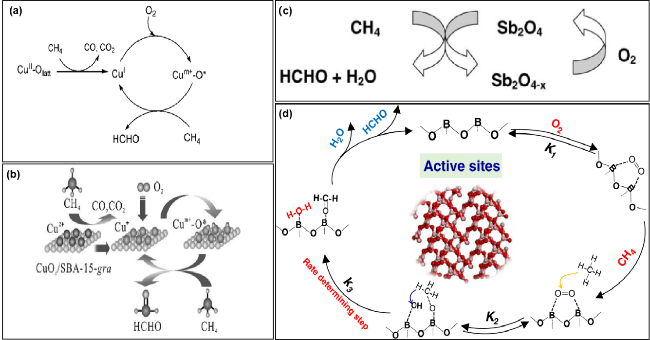
图7 (a) CuOx/SBA-15催化剂上CH4选择性氧化成HCHO的反应机理[58]; (b) CuOx/SBA-15-gra(接枝法)催化剂选择性氧化CH4生成HCHO可能发生的反应机理[163]; (c) α-Sb2O4与Sb2O4-x之间的氧化还原循环[169]; (d) 甲烷在B2O3基催化剂上选择性氧化制甲醛的可行途径示意图[173]Fig.7 (a) Reaction mechanism for selective oxidation of CH4 to HCHO over the CuOx/SBA-15 catalyst[58]; (b) possible reaction mechanism for the selective oxidation of CH4 to HCHO over the CuOx/SBA-15-gra catalysts[163]; (c) catalytic cycle of selective oxidation[169]; (d) schematic diagram of the plausible pathway of partial oxidation of methane to formaldehyde on B2O3 catalysts[173] |
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
( 李孔斋, 王华, 魏永刚, 敖先权, 刘明春. 化学进展, 2008, 20(9): 1306).
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
( 韩春秋, 曹玥晗, 黄川, 吕伟峰, 周莹. 化学进展, 2024, 36(6): 867).
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|
| [24] |
|
| [25] |
|
| [26] |
|
| [27] |
|
| [28] |
|
| [29] |
|
| [30] |
|
| [31] |
|
| [32] |
|
| [33] |
|
| [34] |
|
| [35] |
|
| [36] |
|
| [37] |
|
| [38] |
|
| [39] |
|
| [40] |
|
| [41] |
|
| [42] |
|
| [43] |
|
| [44] |
|
| [45] |
|
| [46] |
|
| [47] |
|
| [48] |
|
| [49] |
|
| [50] |
|
| [51] |
|
| [52] |
|
| [53] |
|
| [54] |
|
| [55] |
|
| [56] |
|
| [57] |
|
| [58] |
|
| [59] |
|
| [60] |
|
| [61] |
|
| [62] |
|
| [63] |
|
| [64] |
|
| [65] |
|
| [66] |
|
| [67] |
|
| [68] |
|
| [69] |
|
| [70] |
|
| [71] |
|
| [72] |
|
| [73] |
|
| [74] |
|
| [75] |
|
| [76] |
|
| [77] |
|
| [78] |
|
| [79] |
|
| [80] |
|
| [81] |
|
| [82] |
|
| [83] |
|
| [84] |
|
| [85] |
|
| [86] |
|
| [87] |
|
| [88] |
|
| [89] |
|
| [90] |
|
| [91] |
|
| [92] |
|
| [93] |
|
| [94] |
|
| [95] |
|
| [96] |
|
| [97] |
|
| [98] |
|
| [99] |
|
| [100] |
|
| [101] |
( 崔湘浩, 魏诠, 李向伟, 马英德. 高等学校化学学报, 1990, 11(10): 1158).
|
| [102] |
|
| [103] |
|
| [104] |
|
| [105] |
|
| [106] |
|
| [107] |
|
| [108] |
|
| [109] |
|
| [110] |
|
| [111] |
|
| [112] |
|
| [113] |
|
| [114] |
|
| [115] |
|
| [116] |
|
| [117] |
|
| [118] |
|
| [119] |
|
| [120] |
|
| [121] |
|
| [122] |
|
| [123] |
|
| [124] |
|
| [125] |
|
| [126] |
|
| [127] |
|
| [128] |
|
| [129] |
|
| [130] |
|
| [131] |
|
| [132] |
|
| [133] |
|
| [134] |
|
| [135] |
|
| [136] |
|
| [137] |
|
| [138] |
|
| [139] |
|
| [140] |
|
| [141] |
|
| [142] |
|
| [143] |
|
| [144] |
|
| [145] |
|
| [146] |
|
| [147] |
|
| [148] |
|
| [149] |
|
| [150] |
|
| [151] |
|
| [152] |
|
| [153] |
|
| [154] |
|
| [155] |
|
| [156] |
|
| [157] |
|
| [158] |
|
| [159] |
|
| [160] |
|
| [161] |
|
| [162] |
|
| [163] |
|
| [164] |
|
| [165] |
|
| [166] |
|
| [167] |
|
| [168] |
|
| [169] |
|
| [170] |
|
| [171] |
|
| [172] |
|
| [173] |
|
| [174] |
|
| [175] |
|
| [176] |
|
| [177] |
( 杨旸, 郑雯, 程党国, 陈丰秋, 詹晓力. 化学进展, 2009, 21(10): 2205).
|
| [178] |
|
| [179] |
|
| [180] |
|
| [181] |
|
/
| 〈 |
|
〉 |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}