
Latent Space Embedding Methods for Chemical Molecules: Principles and Applications
Received date: 2025-03-12
Revised date: 2025-06-19
Online published: 2025-10-15
Supported by
National Natural Science Foundation of China(22272009)
National Natural Science Foundation of China(22203008)
Effective representation of chemical molecules is the key to promoting chemical informatics and new material research and development. In recent years, data-driven molecular representation technology has been developed. Compared with traditional manually designed descriptors and graph structure analysis methods, it can effectively avoid noise and information redundancy, and provide support for efficient and accurate property prediction. Embedding representation has the characteristics of efficient information compression, data representation enhancement and semantic retention, and has been widely used in fields such as deep learning and data mining. Inspired by word embeddings in the field of natural language processing, researchers began to explore the application of similar methods to the construction of the latent space of chemical molecules, and proposed a variety of embedding methods for molecular property prediction and molecular structure generation. This review first elucidates the principles of general embedding technology in machine learning, and then sequentially discusses chemical element latent space representation methods and chemical molecule latent space embedding techniques. By examining the innovative applications of related technologies in natural language processing and graph embedding to molecular embeddings, the review reveals that current molecular embedding methods are gradually evolving towards multimodality, self-supervised learning, and dynamic modeling, and it outlines prospects for future research trends.
1 Introduction
2 Principles of embedding in machine learning
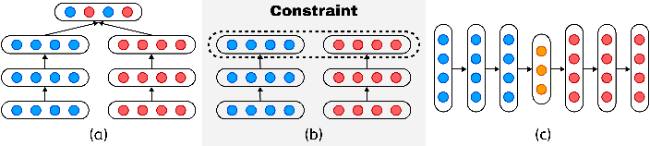
2.1 Word embedding
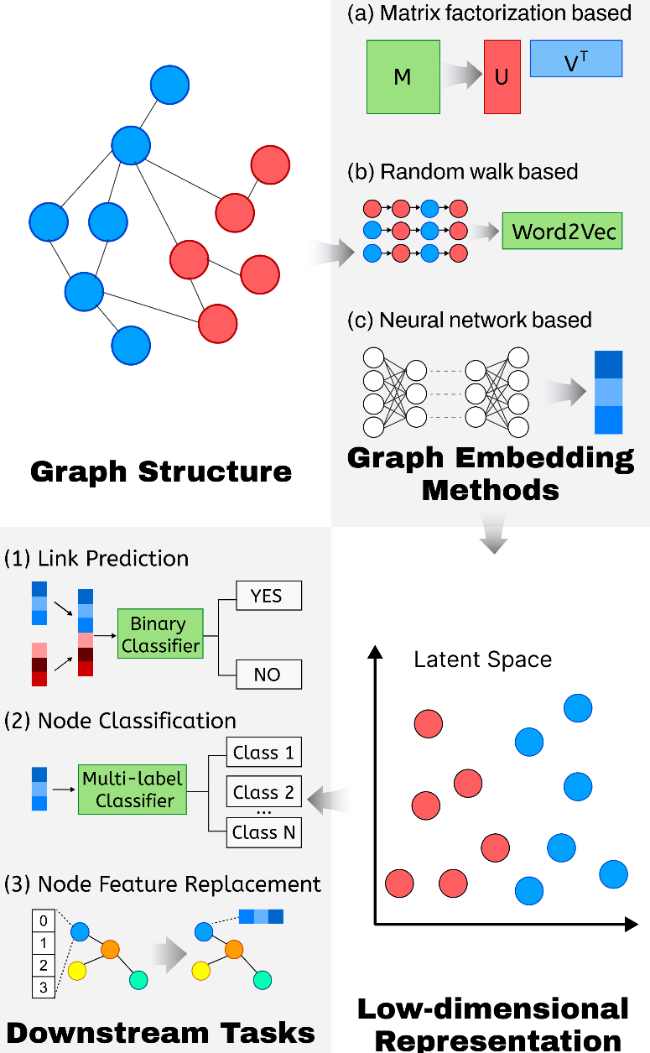
2.2 Graph embedding
2.3 Multimodal embedding
3 Element latent space representation methods
3.1 Attribute-based element representation
3.2 Element representation based on physicochemical knowledge
3.3 Data-driven element embedding
4 Advances in molecular latent space embedding
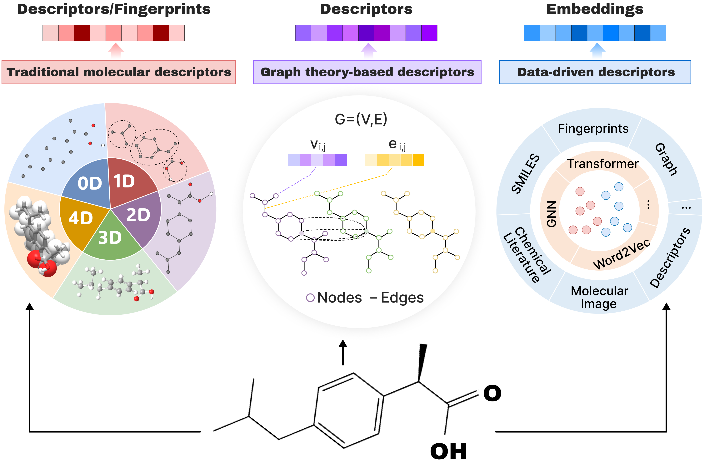
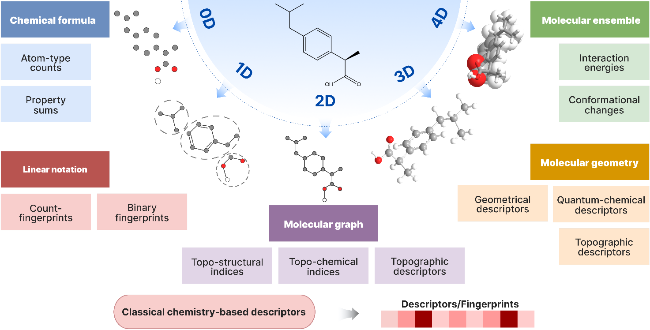
4.1 Traditional chemical feature-based molecular descriptors
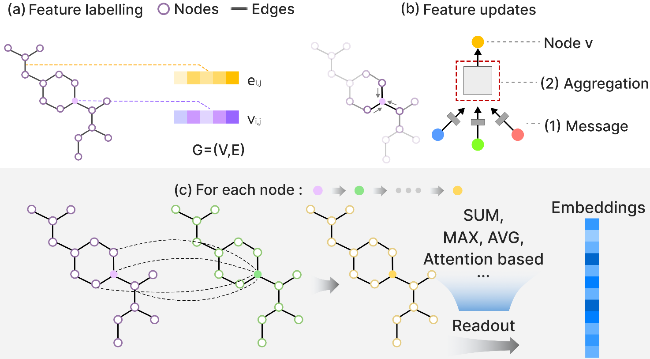
4.2 Graph theory-driven molecular embedding
4.3 Data-driven molecular embedding
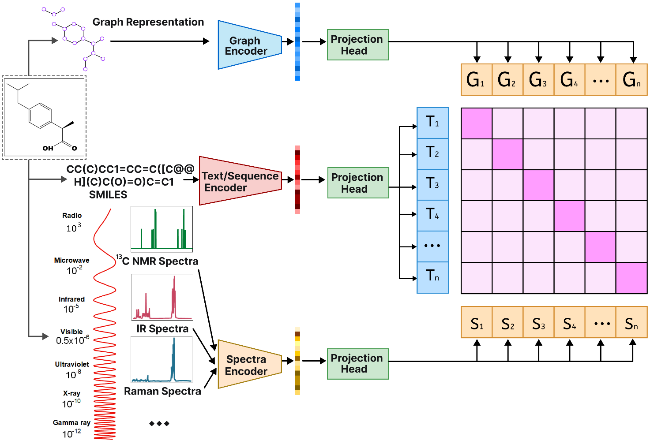
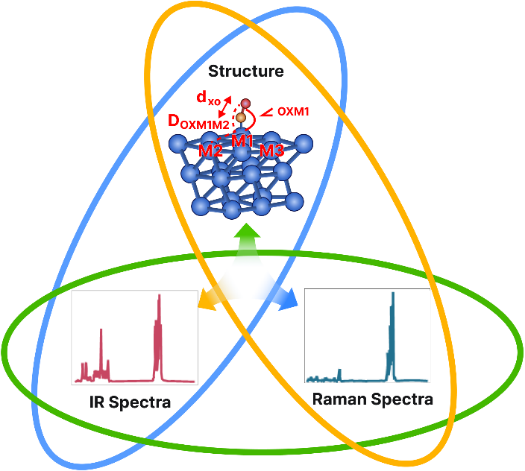
4.4 Multimodal molecular embedding
5 Conclusion and outlook
5.1 Current status and key technology
5.2 Future research prospects
Haotian Chen , Tao Yang , Xiaotong Liu . Latent Space Embedding Methods for Chemical Molecules: Principles and Applications[J]. Progress in Chemistry, 2025 , 37(10) : 1456 -1478 . DOI: 10.7536/PC20250308
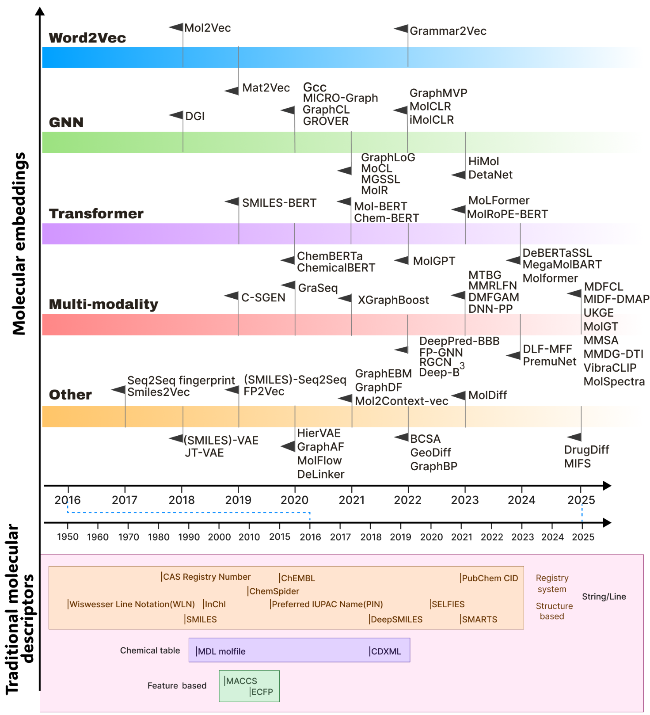
表1 词嵌入方法总结Table 1 Summary of word embedding methods |
| Method | Implementation principles | Ref |
|---|---|---|
| Skip-Gram | Predicts context words based on a given center word. | 6 |
| CBOW | Predicts the center word based on given context words. | 6 |
| GloVe | Constructs a global co-occurrence matrix between words and use the co-occurrence probability to learn word vectors. | 14 |
| FastText | Uses subwords (n-grams) to construct word vectors and effectively handle rare words. | 15 |
| LSA | Reduces the dimension of the word co-occurrence matrix based on singular value decomposition (SVD). | 16 |
| LDA | Models the co-occurrence relationship between documents and topics to obtain the probability distribution of words. | 17 |
| ELMo | Uses a bidirectional Long Short-Term Memory (LSTM) network to obtain contextualized (dynamic) word embeddings. | 18 |
| BERT | Generates context-aware word vectors based on the self-attention mechanism. | 19 |
图2 (a) 基于矩阵分解的方法使用数据特征矩阵通过矩阵分解学习嵌入;(b) 基于随机游走的方法通过随机游走生成节点序列,并应用Word2Vec模型学习嵌入表示;(c) 基于神经网络的方法在不同模型中架构和输入存在差异Fig.2 (a) Matrix factorization-based methods use a data matrix to learn embeddings through factorization. (b) Random walk-based methods generate node sequences via random walks and apply the Word2Vec model to learn embedding representations. (c) Neural network-based methods vary in architecture and input across different models |
表2 GNNs特征选择及聚合信息方法Table 2 GNNs feature selection and information aggregation methods |
| Model | Node features | Edge features | Others | Activation function | Aggregation method | Ref |
|---|---|---|---|---|---|---|
| SchNet | Atomic number | Atomic distances expanded with Gaussian basis functions. | - | Softplus | Uses a filter-generating network (a fully connected neural network) based on interatomic positions to generate filter values. It then performs element-wise multiplication of the neighboring atoms’ atomic representations with these filter values and applies continuous-filter convolution to aggregate information from surrounding atoms. | 62 |
| MEGNet | Atom type, Chirality, Ring sizes, Hybridization, Acceptor, Donor, Aromatic | Bond type, Same ring, Graph distance, Expanded distance | Defines the global state as the average atomic weight and the number of bonds per atom. | Softplus | Employs a multi-layer perceptron with two hidden layers to sequentially update bond attributes, atom attributes, and global state attributes, where the attributes updated in one step are passed to the next update. | 37 |
| DimeNet++ | Atomic number | Atomic distances expanded with radial basis functions (RBF) and bond angles expanded with spherical basis functions (SBF). | - | SiLU | Sums the neighboring message with the message obtained by element-wise multiplying the fully connected layer embedded RBF distance information with the SBF angle information, and then combines this with its own message to complete the message aggregation. | 59 |
| ALIGNN | Electronegativity, Group number, Covalent radius, Valence electrons, First ionization energy, Electron affinity, Block, Atomic volume | Atomic distances expanded with RBF. | Constructs the atomistic line graph from the atomistic graph, where the nodes share latent representations with the bonds in the atomistic graph, and the initial edge features are derived from the RBF expansion of the cosine of bond angles. | SiLU | Performs edge-gated graph convolution[68] on both the atomistic bond graph and the line graph. In the line graph, triplets of atoms and bond features are updated. The newly updated pair features are then propagated to the edges of the direct graph and further updated with the atom features via a second edge-gated graph convolution applied to the direct graph. | 69 |
| eqV2 S DeNS | Atomic number | Atomic distances expanded with RBF and relative position vectors expanded using spherical harmonics (SH). | Utilizes Denoising Non-equilibrium Structures (DeNS)[70] as an auxiliary task by adding noise to the 3D structure, combining it with forces from the original non-equilibrium structure, and predicting the denoised structure. | SiLU | Incorporates relative positions between nodes by rotating the concatenated source and target node features (ensuring equivariance). It decomposes the product of the radial function-generated distance embedding and the rotated node features to compute attention weights and value features, multiplies these, rotates back to the original coordinate system, and finally concatenates features from multiple attention heads to generate new features. | 71 |
表3 数据驱动嵌入方法Table 3 Data-driven embedding approaches |
| Method | Architecture type | Input type | Representation learning strategy | Ref |
|---|---|---|---|---|
| Mol2Vec | Word2Vec | SMILES | Treats compound substructures derived from the Morgan algorithm as "words" and compounds as "sentences", applying the Word2Vec algorithm on a corpus of compounds. | 72 |
| Grammar2Vec | Word2Vec | SMILES | Treats the grammar rules that generate SMILES as "words" and molecules as "sentences", applying the CBOW method on the dataset. | 73 |
| Mat2Vec | Word2Vec | Text (material science abstracts) | Builds a vocabulary of 500000 from 3.3 million scientific abstracts and applies the skip-gram method on the text corpus. | 39 |
| OWL2Vec* | Word2Vec | OWL[74] | Encodes OWL ontology semantics by considering graph structure, lexical information, and logical constructors, implementing an ontology embedding method based on random walks and word embeddings. | 75 |
| Smiles2Vec | RNN | SMILES | Encodes the SMILES string into a fixed-length vector and uses RNN to process the string character by character. | 76 |
| FP2Vec | CNN | Fingerprint | Extracts molecular substructures from SMILES representations, builds a lookup table for the generated fingerprint indices, and maps these indices into trainable embedding vectors. | 77 |
| HiMol | GNN | Molecular graph | Comprises a hierarchical molecular graph neural network and multi-level self-supervised pre-training. It builds a molecular representation learning method based on node-motif-graph hierarchical information, augments graph-level nodes to simulate molecular graph representations, and enables bidirectional transmission of local and global features. | 78 |
| SMILES-BERT | BERT | SMILES | Adjusts the Transformer layer design in BERT and pre-trains the model using a Masked SMILES Recovery task. It also incorporates a quantitative drug similarity prediction task to improve classification performance during fine-tuning. | 79 |
| ChemicalBERT | BERT | Text (chemical texts from PMC abstracts) | Combines ChemicalBERT and AGGCN[80] components to generate high-quality contextual representations and capture syntactic graph information. It merges features from sequential and syntactic graph representations in parallel to predict chemical-protein interaction types. | 81 |
| Mol-BERT | BERT | SMILES | Obtains atomic identifiers with radii 0 and 1 using the Morgan algorithm, uses identifier embeddings as input for pre-training BERT modules, and performs pre-training using only the Masked Language Model (MLM) task. | 82 |
| MolRoPE-BERT | BERT | SMILES | Modifies Mol-BERT by replacing absolute positional encoding with Rotary Positional Encoding (RoPE) to address the insensitivity of the self-attention mechanism to positional information. | 83 |
| Chem-BERT | BERT | SMILES | Designs a matrix embedding layer to learn molecular connectivity. In addition to the MLM task, it adds a quantitative drug similarity prediction task to obtain representations integrating SMILES and chemistry context. | 84 |
| ChemBERTa | RoBERTa | SMILES | Processes SMILES sequences using Byte-Pair Encoding (BPE) tokenization and pre-trains using the MLM task. | 85 |
| DeBERTaSSL | DeBERTa | SMILES | Using SMILES tokenization to represent molecular components, and pre-trains the DeBERTa model with the MLM task. It further combines GCN for self-supervised learning of molecular graph structure information, to capture both sequence and molecular structure information. | 86 |
| MoLFormer | Transformer | SMILES | Combines Rotary Positional Encoding, a linear attention mechanism, and the MLM pre-training method to demonstrate that training on SMILES learns spatial relationships between atoms in a molecule. | 87 |
| Molformer | Transformer | Heterogeneous molecular graph | Constructs heterogeneous molecular graphs by extracting motifs, then uses a Transformer with heterogeneous self-attention to distinguish multi-level node interactions. Incorporates an attentive downsampling algorithm to aggregate informative molecular representations efficiently. | 88 |
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|
| [24] |
|
| [25] |
|
| [26] |
|
| [27] |
|
| [28] |
|
| [29] |
|
| [30] |
|
| [31] |
|
| [32] |
|
| [33] |
|
| [34] |
|
| [35] |
|
| [36] |
|
| [37] |
|
| [38] |
|
| [39] |
|
| [40] |
|
| [41] |
|
| [42] |
|
| [43] |
|
| [44] |
|
| [45] |
|
| [46] |
|
| [47] |
|
| [48] |
|
| [49] |
|
| [50] |
|
| [51] |
|
| [52] |
|
| [53] |
|
| [54] |
|
| [55] |
|
| [56] |
|
| [57] |
|
| [58] |
|
| [59] |
|
| [60] |
|
| [61] |
|
| [62] |
|
| [63] |
|
| [64] |
|
| [65] |
|
| [66] |
|
| [67] |
|
| [68] |
|
| [69] |
|
| [70] |
|
| [71] |
|
| [72] |
|
| [73] |
|
| [74] |
|
| [75] |
|
| [76] |
|
| [77] |
|
| [78] |
|
| [79] |
|
| [80] |
|
| [81] |
|
| [82] |
|
| [83] |
|
| [84] |
|
| [85] |
|
| [86] |
|
| [87] |
|
| [88] |
|
| [89] |
|
| [90] |
|
| [91] |
|
| [92] |
|
| [93] |
|
| [94] |
|
| [95] |
|
| [96] |
|
| [97] |
|
| [98] |
|
| [99] |
|
| [100] |
|
| [101] |
|
| [102] |
|
| [103] |
|
| [104] |
|
| [105] |
|
| [106] |
|
| [107] |
|
| [108] |
|
| [109] |
|
| [110] |
|
| [111] |
|
| [112] |
|
| [113] |
|
| [114] |
|
| [115] |
|
| [116] |
|
| [117] |
|
| [118] |
|
| [119] |
|
| [120] |
|
| [121] |
|
| [122] |
|
| [123] |
|
| [124] |
|
| [125] |
|
| [126] |
|
| [127] |
|
| [128] |
|
| [129] |
|
| [130] |
|
| [131] |
|
| [132] |
|
| [133] |
|
| [134] |
|
| [135] |
|
| [136] |
|
| [137] |
|
| [138] |
|
| [139] |
|
| [140] |
|
| [141] |
|
| [142] |
|
| [143] |
|
| [144] |
|
| [145] |
|
| [146] |
|
| [147] |
|
| [148] |
|
| [149] |
|
| [150] |
|
| [151] |
|
| [152] |
|
| [153] |
|
| [154] |
|
| [155] |
|
| [156] |
|
| [157] |
|
| [158] |
|
| [159] |
|
| [160] |
|
| [161] |
|
| [162] |
|
| [163] |
|
| [164] |
|
| [165] |
|
| [166] |
|
| [167] |
|
| [168] |
|
| [169] |
|
| [170] |
|
| [171] |
|
| [172] |
|
| [173] |
|
| [174] |
|
| [175] |
|
| [176] |
|
| [177] |
|
| [178] |
|
| [179] |
|
| [180] |
|
| [181] |
|
| [182] |
|
| [183] |
|
| [184] |
|
| [185] |
|
| [186] |
|
| [187] |
|
| [188] |
|
| [189] |
|
| [190] |
EdwinChacko, RudraSondhi,
|
| [191] |
|
| [192] |
|
| [193] |
|
| [194] |
|
| [195] |
|
/
| 〈 |
|
〉 |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}