
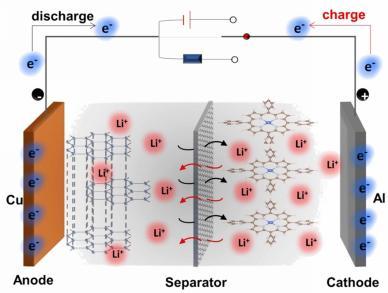
Organic Electrode Materials: Classification and Typical Modification Applications in Metal-Ion Batteries
Received date: 2025-03-04
Revised date: 2025-04-24
Online published: 2025-10-13
Supported by
National Natural Science Foundation of China(52371248)
Guangdong Basic and Applied Basic Research Foundation(2023A1515010905)
Guangdong Basic and Applied Basic Research Foundation(2024A1515140004)
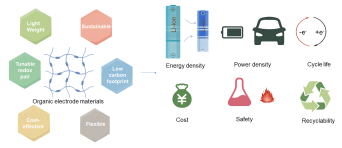
The pursuit of green and sustainable development has become a global consensus, also prompting the vigorous exploration of novel electrode materials within the realm of battery technology. As a result, organic electrode materials have garnered widespread attention. Compared to traditional electrode materials, organic electrode materials offer advantages such as high structural flexibility, tunable electrical properties, and being environmentally friendly and low-cost. These benefits make them versatile in battery applications. However, during the application process, issues such as the molecular structure and conjugated system of the material can lead to difficulties in electron transport, resulting in poor conductivity. Additionally, due to their chemical structure and polarity, many organic electrode materials have high solubility in electrolytes, causing loss of active material and leading to poor cycling stability and capacity fade in batteries. Therefore, it is necessary to modify the molecular structure design of the material. This review provides an in-depth analysis of the development of organic electrode materials in the field of batteries. Comparing them with inorganic electrode materials, it reveals their unique application advantages. It also elaborates on the electrochemical mechanisms of different types of organic electrode materials and explores in detail the applications of various organic electrode materials in different metal-ion batteries and the further improvement measures. The review focuses on modifying various organic electrode materials, such as carbonyl compounds, organic sulfides, and organic radicals, for their applications in metal-ion batteries. This is achieved through perspectives like molecular design, polymerization, compositing with different materials, and regulating micro/nanostructures. These modifications aim to enhance conductivity and cycling stability, thereby realizing the long-life development of batteries. Finally, the review looks forward to the future development of organic electrode materials, hoping that by summarizing different modification measures and controlling various optimization methods, electrode materials with higher performance and fewer defects can be developed. It is believed that through continuous summarization and improvement, organic electrode materials can achieve higher performance upgrades, make greater breakthroughs in future applications, reach more diverse application levels, and contribute to green and sustainable development.
1 Introduction
2 OEMs vs IEMs
3 Electrochemical mechanism
4 Types of OEMs
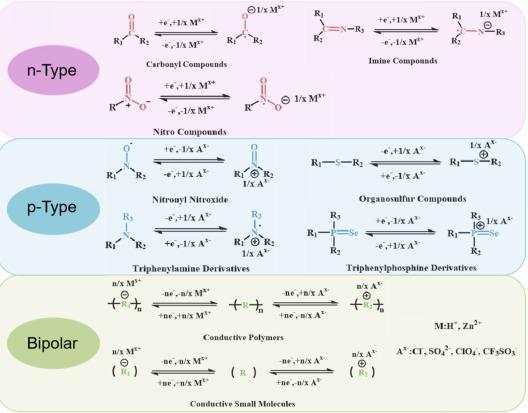
4.1 N-type OEMs
4.2 P-type OEMs
4.3 Bipolar OEMs
5 Structure, classification, and modification
5.1 Carbonyl compound
5.2 Organic sulfide
5.3 Heterocyclic compound
5.4 Organic radical
5.5 Other OEMs
6 Conclusion and outlook
Mengyuan Hao , Qing Meng , Yachao Yan , Yingzhi Chen , Jiantao Wang , Luning Wang . Organic Electrode Materials: Classification and Typical Modification Applications in Metal-Ion Batteries[J]. Progress in Chemistry, 2025 , 37(10) : 1479 -1512 . DOI: 10.7536/PC20250304
表1 有机电极材料在不同类型电池中的应用Table 1 Applications of organic electrode materials in different types of batteries |
| Battery category | Application | ref |
|---|---|---|
| Aluminum-ion batteries (AIB) | Synergistic oxidation-reduction sites; combined with organic frameworks; polymerization of various organic compounds | 39~43 |
| Sodium-ion batteries (SIBs) | Aggregation of various organic compounds; preparation of organic linking agents | 44~47 |
| Potassium-ion batteries (KIBs) | Optimize the composition of organic electrode materials; construct multi-component polymers | 48~51 |
| Calcium-ion batteries (CIBs) | Direct application; constructing organic frameworks; composite application | 52~55 |
表2 有机电极材料与无机电极材料的比较Table 2 Comparison of organic and inorganic electrode materials |
| Electrode materials category | Advantages | Challenges | ref |
|---|---|---|---|
| Organic electrode materials | Sustainability; tunability of molecular structure; the molecular skeleton is soft; environmentally friendly and low-carbon; lightweight | Low conductivity; unclear charging and discharging mechanism; not balancing high specific capacity and long stable cycling performance | 64~69 |
| Inorganic electrode materials | Usually insoluble in organic solvents; clear charging and discharging mechanism | Rigid structure; volume changes significantly during charging and discharging; unsustainable; environmental pollution | 70~75 |
表3 有机电极材料案例总结Table 3 Summary of organic electrode material cases |
| Cassification | Molecular structure | Modification strategy | Specific capacity (mAh·g-1) | Cyclic conditions | Capacity Retention (mAh·g-1) | Improved capacity performance after cycling | Battery type | ref |
|---|---|---|---|---|---|---|---|---|
| Quinones and their derivatives | 2,2′-(1,4-phenylenedithio) bis(1,4-naphthoquinone) (1,4-PNQ)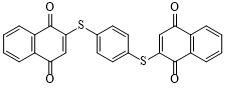 | Molecular design: insertion of benzenedithiol(1,4-BDT) between two NQ molecules | 231 (at 1 C) | 100 cycles at 1 C | 185/80% | 1.8 times higher than NQ (103 mAh·g-1) | Lithium-ion battery | 101 |
| Quinones | 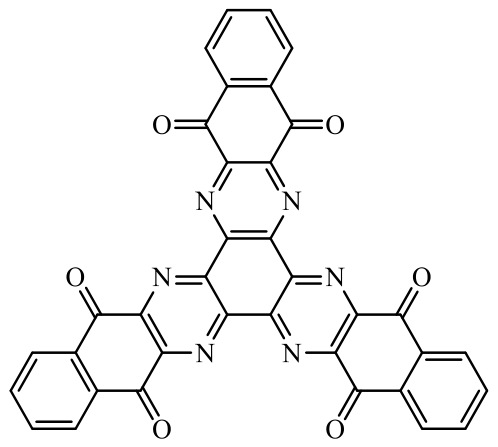 Hexaazatrianthranylene (HATAQ) Hexaazatrianthranylene (HATAQ) | Molecular design: the conjugated quinone moiety is introduced into the nucleus of an electron-deficient hexazatribenzene derivative | 426 (at 200 mA·g-1) | 100 cycles at 0.4 C | 376/86.4% | 1.2 times higher than the general organic cathode small molecule materials | Lithium-ion battery | 102 |
| Quinones/pyrazine hybrid structure |  5,7,12,14-Tetraaza-6,13- Pentacenequinone (TAPQ) | Molecular design: integrating electroactive quinone and pyrazine groups in small molecules | 270 (at 50 mA·g-1) | 250 cycles at 50 mA·g-1 | 248/92% | - | Aqueous Rechargeable Zinc Batteries | 104 |
| Carboxylic compounds | 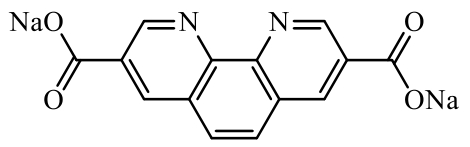 sodium 1,10-phenanthroline-3,8-dicarboxylate (S-PD) sodium 1,10-phenanthroline-3,8-dicarboxylate (S-PD) | Molecular design: incorporating two Na+ ions into the organic small molecule of phenanthroline-3,8-dicarboxylate | 252 (at 50 mA·g-1) | 250 cycles at 2 C | 126/50% | - | Sodium-Ion Battery | 112 |
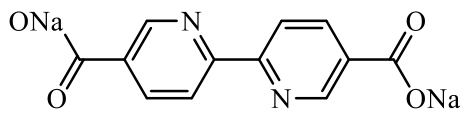 disodium 2,2'-bipyridine-5,5'- disodium 2,2'-bipyridine-5,5'-dicarboxylate (2255-Na) | Molecular design: incorporating nitrogen atoms into conjugated structures and regulating the position of carboxylate groups on aromatic rings | 210 (at 50 mA·g-1) | 500 cycles at 50 mA·g-1 | 178/85% | 1.32 times higher than disodium 4,4’-biphenyl dicarboxylate (BPDC-Na) | Sodium-Ion Battery | 113 | |
| Imine |  2,9-dioctylperylene-3,4,9,10-tetracarboxylic diimide (2PDI) | Molecular design: reversible co-insertion Zn2+/H+ at the carbonyl site | 72.9 (at 100 mA·g-1) | 500 cycles at 100 mA·g-1 | 70.1/96.2% | 4.56 times higher than PTCDA//Zn(21.1%) | Aqueous Zinc-Organic Batteries | 117 |
| Carbonyl polyquinone imine |  Polyquinoneimine Sandwiched by Graphene (PQI@Gr) Polyquinoneimine Sandwiched by Graphene (PQI@Gr) | Polymerization and composite: the polymerization of dianhydride and anthraquinone combined with graphene oxide | 205 (at 0.1 A·g-1) | 10000 cycles at 5 A·g-1 | 87.7/73% | 2.34 times higher than PQI | Lithium-Ion Battery | 119 |
| Organic sulfur compounds | 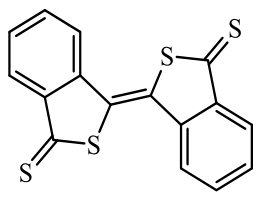 Benzo[c]thiophen-1(3H)-thione (DPTTO) | Molecular design: replacing the oxygen atom in benzo[c]thiophene-1(3H)-one (DPTO) with sulfur atom | 162 (at 50 mA·g-1) | 200 cycles at 50 mA·g-1 | 122/75% | The initial retention rate is 4.26 times that of DPTO | Sulfur-rich All-Organic Lithium-Ion Batteries | 125 |
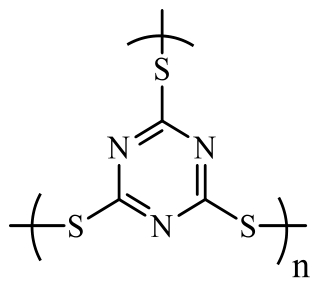 Poly(trithiocyanuric acid (PTTCA) | Composite carbon nanotubes PTTCA@CNT | 468 (at 50 mA·g-1) | 100 cycles at 50 mA·g-1 | 388/83% | Capacity retention rate increased 2.1 times compared to bare PTTCA | All-Solid-State Lithium Battery | 126 | |
| Heterocyclic compound: polythiophene | 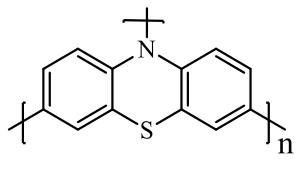 polyphenothiazine (PPTZ) polyphenothiazine (PPTZ) | Oxidative polymerization | 157 (at 50 mA·g-1) | 100 cycles at 50 mA·g-1 | 127/81% | 1.37 times higher than phenothiazine monomer (PTZ) | Potassium-basedBattery | 148 |
| Heterocyclic compound: phenazine | 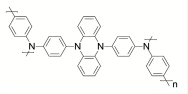 Poly(Hyperbranched dihydrophenazine diphenylamine) (PDPAPZ) Poly(Hyperbranched dihydrophenazine diphenylamine) (PDPAPZ) | Cross-coupling polymerization | 101 (at 5 A·g-1) | 100 cycles at 5 A·g-1 | 87/86% | The specific capacity is comparable to that of the best ultra-fast charging polyamine cathode material PDPPD(84mAh·g-1). | Potassium-based Battery | 149 |
| polymer | 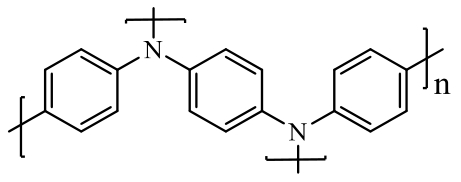 poly(N,N′-diphenyl-p- poly(N,N′-diphenyl-p-phenylenediamine) (PDPPD) | Cross-coupling polymerization | 102 (at 50 mA·g-1) | 500 cycles at 50 mA·g-1 | 79/77% | - | Lithium-based Battery | 150 |
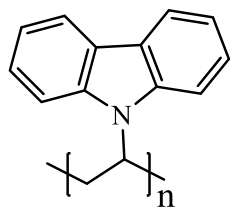 poly(N-vinylcarbazole) (PVK) | polymer | 120 (at 20 mA·g-1) | 400 cycles at 500 mA·g-1 | 84/70% | - | Potassium-basedBattery | 151 | |
| Covalent organic frameworks (COFs) | 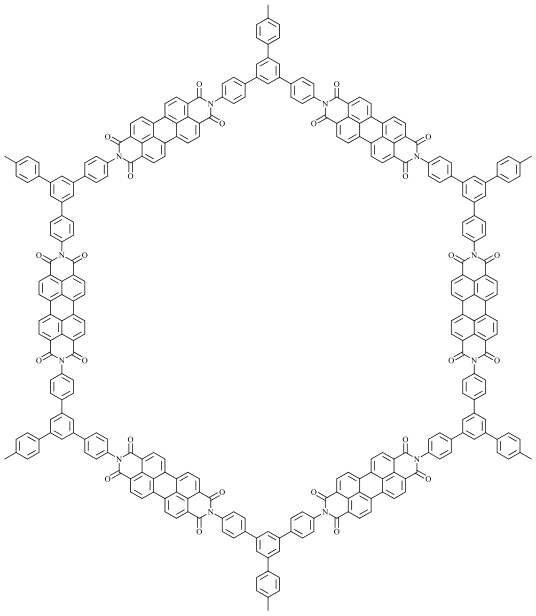 Perylene-based Imide Covalent Organic Framework (Pe-PICOF) Perylene-based Imide Covalent Organic Framework (Pe-PICOF) | Molecular design: adjusting the size of the imide conjugated units through different coordinating ions (Be, Na, Pe) | 91 (at 50 mA·g-1) | 100 cycles at 50 mA·g-1 | 87/96% | 1.71 times higher than Be-PICOF | Lithium-Ion Batteries | 146 |
| Other electrode materials | 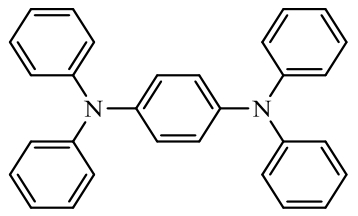 1,4 bis(diphenylamino)benzene (BDB) | Composite: Cellulose Nanocrystal Film | 112 (at 390 mA·g-1) | 500 cycles at 390 mA·g-1 | 84/75% | - | Zinc-based Battery | 152 |
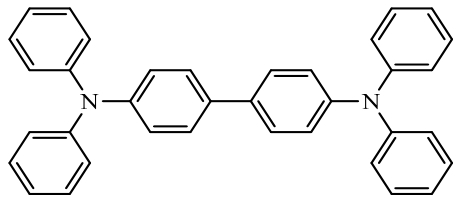 N,N,N′,N′- tetraphenyl-1,1-biphenyl-4,4′-diamine (TPB) | Injecting and transporting holes into TPB for storing PF6- | 77 (at 50 mA·g-1) | 450 cycles at 50 mA·g-1 | 47/61% | - | Lithium-based Battery | 153 |
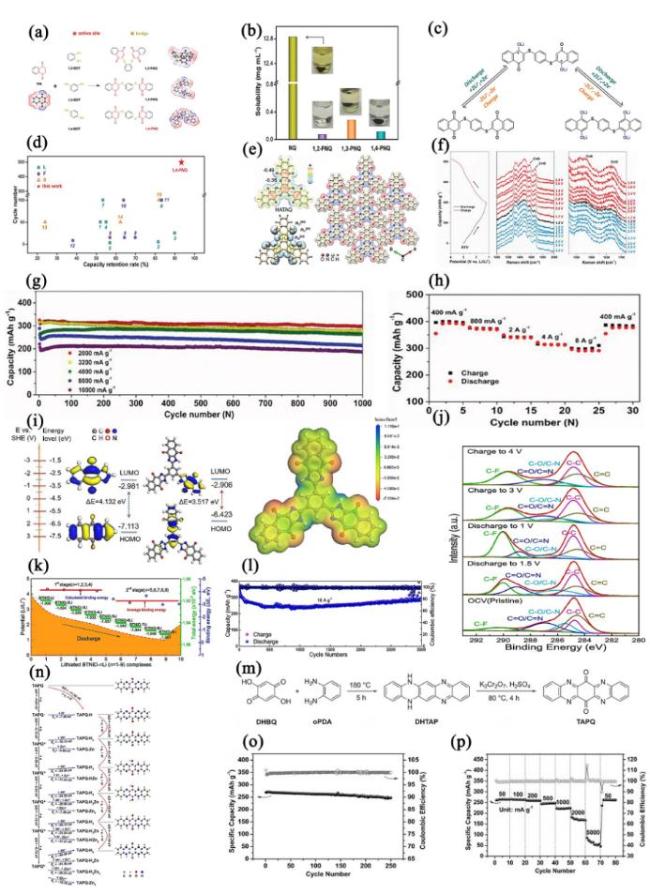
图3 (a) NQ、1,2-PNQ、1,3-PNQ和1,4-PNQ的分子设计类型及其静电势图; (b) NQ、1,2-PNQ、1,3-PNQ和1,4-PNQ电极在G4电解液中浸泡3天后的溶解性和数码照片; (c) 1,4-PNQ的电化学氧化还原反应; (d) 1,4-PNQ和其他醌基电极的容量保持率比较[101]; (e) HATAQ(0.005 e/a03等值面)的化学结构和静电势图; HATAQ分子通过C―H···O键连接形成的二维层状单晶结构; HATAQ分子的σ型孤对电子(n(σ))和π型孤对电子(n(π)); (f) HATAQ电极在不同充电状态下的原位拉曼光谱及在1.2~3.9 V电压范围内200 mA·g-1电流密度下的充放电曲线; (g) HATAQ在高倍率下的容量保持率; (h) HATAQ在不同电流密度下的倍率性能[102]; (i) IMNQ、BDNIB和BTNID的计算轨迹图; 由DFT计算得到的BTNID分子静电势图; (j) BTNID中非配位C元素的高分辨XPS谱图; (k) BTNID电极的锂离子传输路径; (l) BTNID//Li电池在10 A·g-1下循环3000次的容量保持率和库仑效率[103]; (m) TAPQ的合成路线; (n) TAPQ-HxZny的结合能; TAPQ相关分子间的吉布斯自由能差; TAPQ分子及其可能还原产物的最优几何结构; (o) 0.5~1.6 V电压范围内、50 mA·g-1电流密度下TAPQ在1 M ZnSO4电解液中循环250次的长循环性能, (p) TAPQ在不同电流密度下的倍率性能[104]Fig.3 (a) Molecular design types and electrostatic potential maps of NQ, 1,2-PNQ, 1,3-PNQ, and 1,4-PNQ; (b) solubility and digital photos of NQ, 1,2-PNQ, 1,3-PNQ, and 1,4-PNQ electrodes after 3-day immersion in G4 electrolyte; (c) electrochemical redox reactions of 1,4-PNQ; (d) capacity retention comparison of 1,4-PNQ and other quinone-based electrodes. Adapted with permission[101]. (e) Chemical structure and electrostatic potential diagram of HATAQ(0.005 e/a03 isovalue); 2D layered single crystal structure of HATAQ formed by C—H···O bonds;σ-type (n(σ)) and π-type lone pair electrons (n(π)) of HATAQ molecule; (f) in situ Raman spectra of HATAQ electrodes at different charging states, and charge-discharge curves of 200 mA·g-1 within 1.2~3.9 V voltage range; (g) capacity retention rate of HATAQ at high rates; (h) magnification performance of HATAQ at different current densities. Adapted with permission[102]. (i) Computed track maps of IMNQ, BDNIB, and BTNID; Electrostatic potential map of the BTNID molecule obtained from DFT calculations; (j) high-resolution XPS spectra of the off-location C element of BTNID; (k) lithium path of BTNID electrode; (l) capacity retention rate and coulomb efficiency over 3000 cycles at 10 A·g-1 of BTNID//Li battery. Adapted with permission[103]. (m) TAPQ synthesis route; (n) binding energy of TAPQ-HxZny, Gibbs free energy difference among TAPQ related molecules; optimal geometry of TAPQ molecules and possible reduction products; (o) long cycle performance of TAPQ (50 mA·g-1, 250 cycles) with 0.5~1.6 V voltage range in 1 M ZnSO4 electrolyte; (p) magnification performance at different current rates of TAPQ. Adapted with permission[104] |
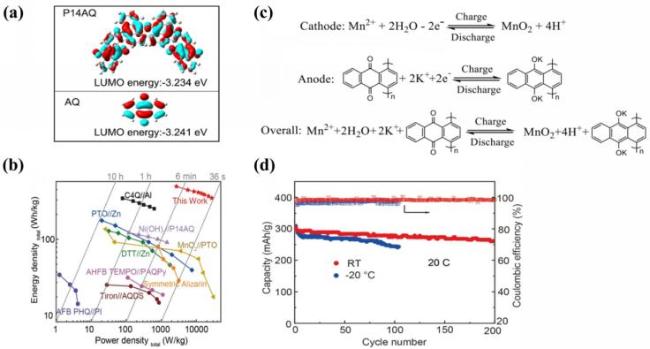
图4 (a) 真空条件下P14AQ和AQ的最低未占据分子轨道(LUMO)能量及轨道分布模式; (b) 与其他已报道的使用醌基电极的水系能量存储系统的Ragone图对比; (c) 自制碱酸混合P14AQ//MnO2电池的氧化还原反应; (d) 20 C下P14AQ//MnO2电池在室温和-20 ℃下的循环性能[105]Fig.4 (a) LUMO energy and orbital distribution of P14AQ and AQ in vacuum; (b) comparison of Ragone plots with other reported aqueous energy storage systems utilizing quinone-based electrodes; (c) redox reactions of fabricated alkali-acid hybrid P14AQ//MnO2 battery; (d) cycle performance of P14AQ//MnO2 battery at room temperature and -20 ℃ at 20 C. Adapted with permission[105] |
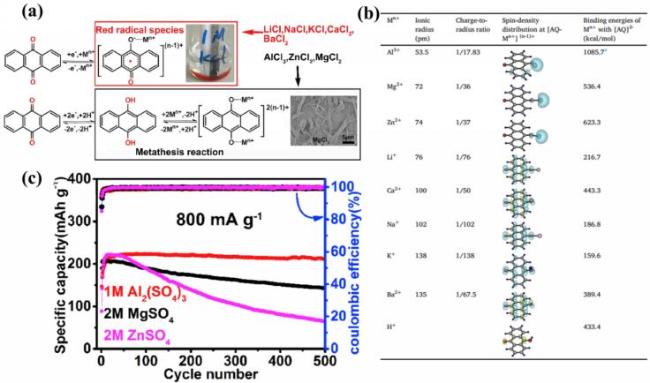
图5 (a) AQ在不同水系金属离子电解液中的氧化还原机制; (b) 不同金属离子的离子半径、[AQ-Mn+](n-1)+的自旋密度分布以及Mn+与[AQ]2-的结合能比较; (c) 800 mA·g-1下AQ的循环稳定性[106]Fig.5 (a) Redox mechanism of AQ in metal ion electrolytes of different water systems; (b) different metal ions of the ionic radius, [AQ-Mn+](n-1)+ spin density distribution and Mn+ and [AQ]2- compare the binding energy; (c) the cyclic stability of AQ at 800 mA·g-1. Adapted with permission[106] |
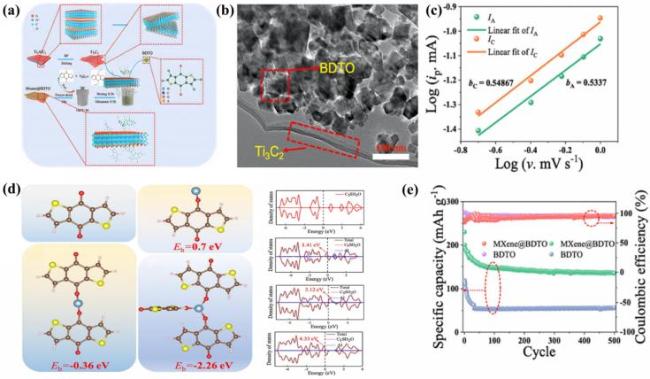
图6 (a) MXene@BDTO的制备过程图; (b) MXene@BDTO的透射电子显微镜图; (c) 氧化峰(1.55 V)和还原峰(1.14 V)处的扫描速率对数值与电流对数值之间的关系; (d) BDTO以不同结合形式与Al3+的吸附能及分子与结合不同数量Al3+时的态密度; (e) MXene@BDTO和BDTO的长循环性能[107]Fig.6 (a) Preparation process diagram of MXene@BDTO; (b) TEM images of MXene@BDTO; (c) correlation between log(scan rate) and log(current) at 1.55 V oxidation peak and 1.14 V reduction peak; (d) adsorption energy of BDTO in different configurations with Al3+ and density of states for the molecule binding different quantities of Al3+; (e) long cycle of MXene@BDTO and BDTO[107]. Adapted with permission |
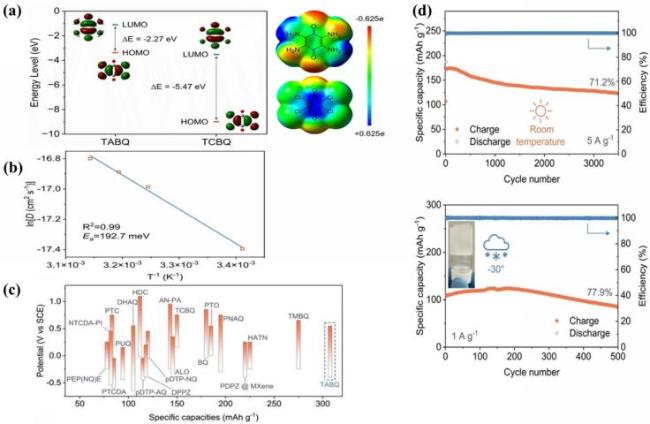
图7 (a) TABQ和TCBQ分子的轨道能量和分子静电势分布; (b) TABQ的ln(DH)与T-1的关系图; (c) 与近期报道的用于水系质子电池的有机电极材料的性能对比; (d) TABQ//TCBQ全有机全电池在室温和-30 ℃下的倍率性能和循环性能[108]Fig.7 (a) Orbital energy and electrostatic potential distribution of TABQ and TCBQ molecules; (b) graph of ln(DH) against T-1 of TABQ; (c) performance comparison with recently reported organic electrode materials in aqueous proton batteries; (d) TABQ//TCBQ all-organic all-battery rate capability and cycle stability at room temperature and -30 ℃. Adapted with permission[108] |
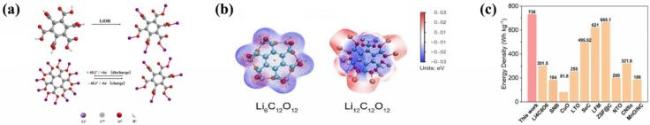
图8 (a) Li6C12O12的合成过程图及其作为锂离子电池电极的储锂机制; (b) Li6C12O12和Li12C12O12的静电势分布; (c) Li6C12O12与其他已报道的锂离子电池负极材料的能量密度对比[110]Fig.8 (a) Diagram of the synthesis diagram of Li6C12O12 and and its lithium storage mechanism as an electrode in lithium-ion batteries; (b) electrostatic potential distribution of Li6C12O12 and Li12C12O12; (c) energy density of Li6C12O12 compared to reported lithium ion battery anode materials. Adapted with permission[110] |
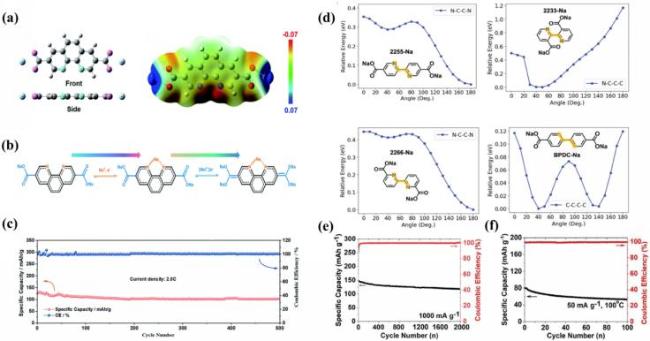
图9 (a) S-PD分子的正视图和侧视图及其静电势分布; (b) S-PD的可逆电化学氧化还原机制; (c) S-PD在2 C倍率下的长期循环稳定性[112]; (d) 在wB97X-D/6-311++G(d,p)理论水平下计算的四个单体的X-C-C′-X二面角的势能面; (e) 2255-Na在钠离子电池中1 A·g-1下的循环寿命和库仑效率; (f) PANI||2255-Na全电池在100 ℃下的循环寿命和库仑效率[113]Fig.9 (a) Front and side view of S-PD molecule and electrostatic potential distribution; (b) electrochemically reversible redox mechanism of S-PD; (c) long-term cyclic stability of S-PD at 2 C. Adapted with permission[112]. (d) potential energy surfaces of four monomers calculated for X-C-C′-X dihedral angles at wB97X-D/6-311++G(d,p) level; (e) cycle performance and coulomb efficiency of 2255-Na in sodium-ion batteries at 1 A·g-1; (f) cycle performance and Coulomb efficiency of PANI||2255-Na full battery at 100 ℃. Adapted with permission[113] |
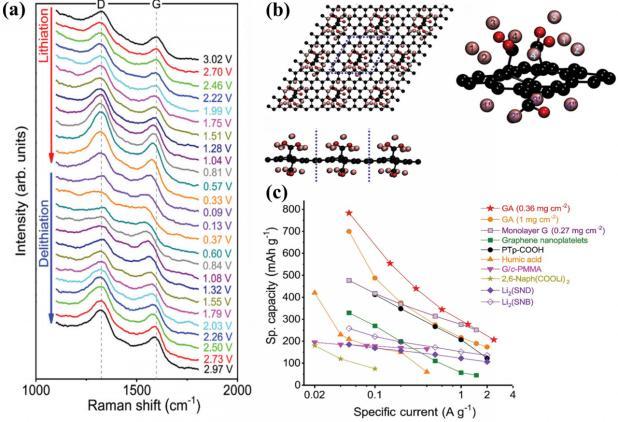
图10 (a) GA负极在不同电位下的原位拉曼光谱; (b) 锂化GA的顶视图、侧视图和放大视图; (c) 有机材料(PTp-COOH、腐殖酸、G/c-PMMA、2,6-Naph(COOLi)2、Li2(SND)、Li2(SNB))以及使用商业石墨烯(单层和纳米片)制备的电极的性能对比[114]Fig.10 (a) In situ Raman spectroscopy of GA negative electrode at varying potentials; (b) top, side and enlarged view of lithiated GA; (c) comparison of the properties of organic materials (PTp-COOH,humic acid, G/c-PMMA, 2,6-Naph(COOLi)2, Li2(SND), Li2(SNB)) and electrodes prepared using commercial graphene (single layer and nanosheet)). Adapted with permission[114] |
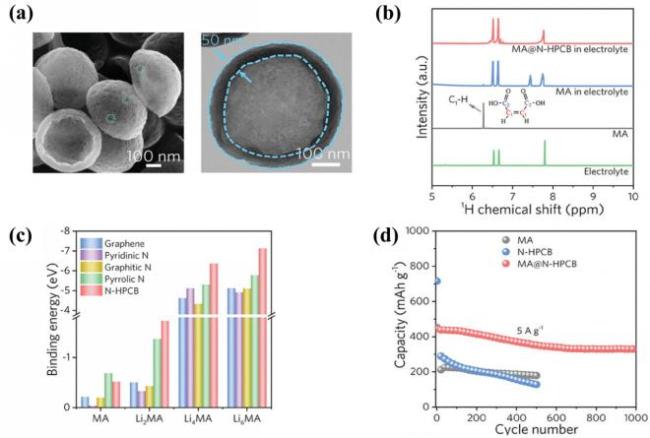
图11 (a) N-HPCB的扫描与透射电子显微镜图像; (b) 浸泡在电解液中的MA和MA@N-HPCB的1H核磁共振谱图; (c) 不同物质在LixMA(x=0,2,4,6)上的结合能; (d) MA、N-HPCB和MA@N-HPCB电极在5 A·g-1下的长期循环性能[115]Fig.11 (a) Scanning and transmission electron microscope images of N-HPCB; (b) 1H NMR spectra of MA and MA@N-HPCB soaked electrolyte; (c) binding energy of different substances on LixMA (x=0,2,4,6); (d) long-cycle performance of MA, N-HPCB and MA@N-HPCB electrodes at 5 A·g-1. Adapted with permission[115] |
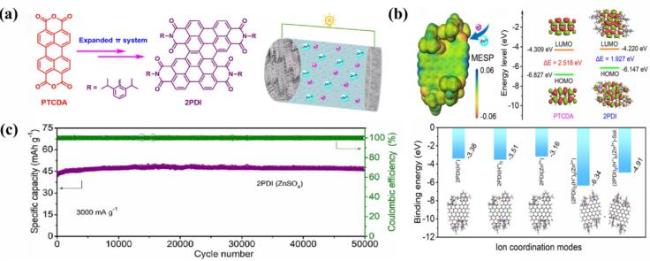
图12 (a) PTCDA和2PDI的分子结构及基于2PDI的水系锌离子电池的示意图; (b) 2PDI分子的范德华表面静电势分布、PTCDA分子的前线分子轨道能量以及分子吸附H+/Zn2+离子的几何构型和计算的结合能; (c) 2PDI电池在3000 mA·g-1下的循环性能和库仑效率[117]Fig.12 (a) Molecular structure of PTCDA and 2PDI and schematic diagram of water zinc-ion battery based on 2PDI; (b) electrostatic potential distribution on the van der Waals surface of 2PDI, molecular orbital energy levels of PTCDA, and geometries and binding energies of H+/Zn2+ ion adsorption on the molecules; (c) cycle stability and coulomb efficiency of 2PDI battery at 3000 mA·g-1. Adapted with permission[117] |
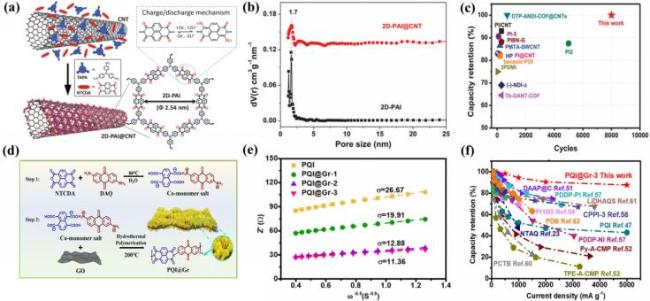
图13 (a) 晶体2D-PAI@CNT合成及能量存储过程示意图; (b) 2D-PAI和2D-PAI@CNT的孔径分布; (c) 与其他聚酰亚胺正极材料的循环性能比较[118]; (d) PQI@Gr的合成路线; (e) PQI和PQI@Gr电极的复阻抗实部与频率平方根(ω-0.5)之间的线性关系; (f) 已报道的PQI@Gr-3以及羰基聚合物正极材料的放大特性[119]Fig.13 (a) Diagram of crystal 2D-PAI@CNT synthesis and energy storage mechanism; (b) aperture distributions of 2D-PAI and 2D-PAI@CNT; (c) cycle stability comparison against other polyimide cathode materials. Adapted with permission[118]. (d) Preparation pathway of PQI@Gr; (e) linear relationship between the real part of the complex impedance and the square root of the frequency (ω-0.5) of PQI and PQI@Gr electrodes; (f) scalability features of reported PQI@Gr-3 and carbonyl polymer cathode materials. Adapted with permission[119] |
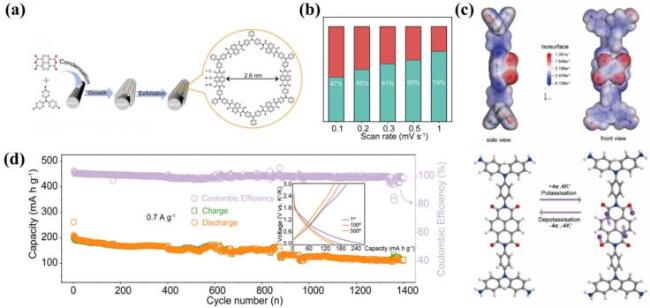
图14 (a) P-COF@SWCNT的合成路线; (b) P-COF@SWCNT在不同扫描速率下电容控制贡献的比例; (c) P-COF插层K+的电荷密度和K+存储机制的示意图; (d) P-COF@SWCNT在0.7 A·g-1下的循环性能以及第1次、第100次和第500次的充放电曲线(示意图)[123]Fig.14 (a) Preparation method of P-COF@SWCNT; (b) P-COF@SWCNT Ratio of capacitance control contribution at various scan rates; (c) schematic diagram of charge density and K+ storage mechanism of P-COF intercalation K+; (d) cycle stability of P-COF@SWCNT at 0.7 A·g-1 with charge-discharge profiles of the first, 100th, and 500th (illustration). Adapted with permission[123] |
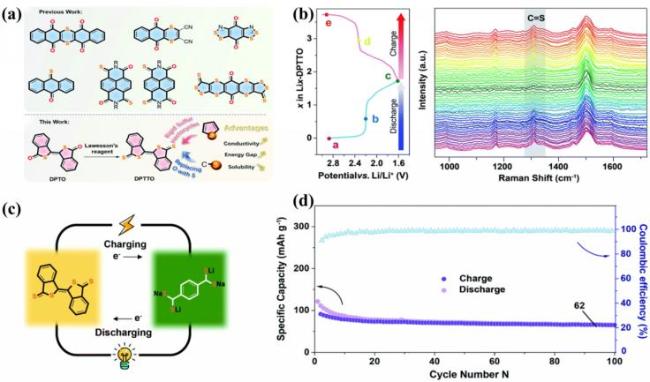
图15 (a) 含硫正极材料及DPTO的硫化过程; (b) DPTTO在20 mA·g-1下的充放电曲线和原位拉曼光谱; (c) 全有机扣式电池的示意图; (d) 电池在100 mA·g-1下的循环性能[125]Fig.15 (a) Sulfur-based cathode materials and sulfidation process of DPTO; (b) charge-discharge curve and in-situ Raman spectrum of DPTTO at 20 mA·g-1; (c) illustration of an all-organic button battery; (d) cycle stability of the battery at 100 mA·g-1. Adapted with permission[125] |
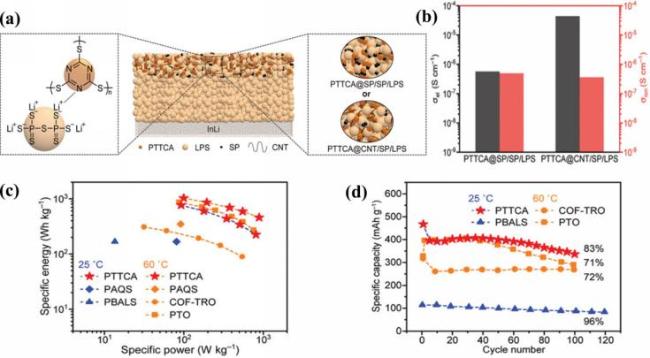
图16 (a) 基于LPS电解质和PTTCA正极的全固态锂金属电池示意图(中间)、正极中LPS与PTTCA的相互作用(左侧)以及使用两种复合材料作为正极的结构(右侧); (b) PTTCA@SP/SP/LPS和PTTCA@CNT/SP/LPS正极颗粒的电子与离子导电性柱状图; (c) 各种阴极材料的Ragone图; (d) 基于硫化物电解质的全固态锂电池中PTTCA与其他有机正极材料的循环性能对比[126]Fig.16 (a) Diagram of an all-solid-state lithium metal battery featuring LPS electrolyte and PTTCA positive electrode (center), the interaction of LPS and PTTCA in the positive electrode (left) and the structure using two composite materials as the positive electrode (right); (b) bar charts illustrating the electronic and ionic conductivity of PTTCA@SP/SP/LPS and PTTCA@CNT/SP/LPS cathode particles; (c) Ragone chart for different cathode materials; (d) comparison of cycle stability of PTTCA and other organic cathode materials in all-solid-state lithium batteries based on sulfide electrolyte. Adapted with permission[126] |
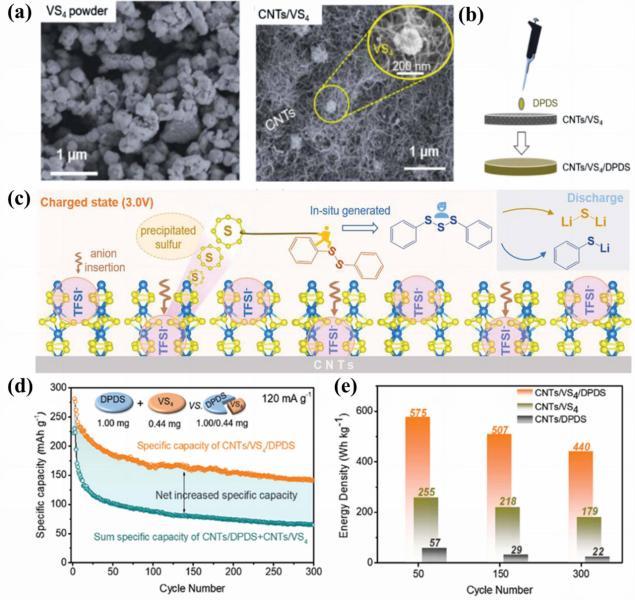
图17 (a) 合成的VS4及CNT/VS4复合物的SEM图像; (b) DPDS电解液添加到CNT/VS4中的示意图; (c) CNTs/VS4/DPDS充放电机制示意图; (d) CNT/VS4/DPDS比容量与CNT/DPDS和CNT/VS4总比容量之间的比较; (e) 三种复合材料在第50、150和300次循环时的质量能量密度的比较[127]Fig.17 (a) The SEM images of the synthesized VS4 and the CNT/VS4; (b) diagram of adding DPDS electrolyte to CNT/VS4; (c) diagram of the charge-discharge mechanism for CNTs/VS4/DPDS; (d) comparison between the specific capacity of CNT/VS4/DPDS and the sum specific capacity of CNT/DPDS and CNT/VS4; (e) comparison of the mass energy density of the three composite materials at the 50th, 150th, and 300th cycles[127] |
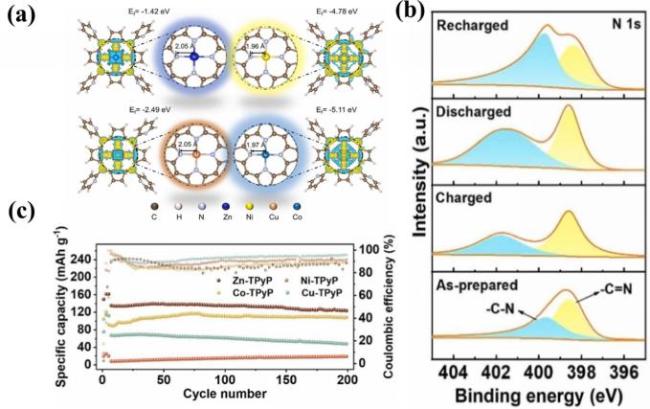
图18 (a) Zn-TPyP、Ni-TPyP、Cu-TPyP和Co-TPyP的原子结构和电荷密度的差异; (b) Zn-TPyP的N 1s XPS谱图; (c) 由不同金属卟啉构建的钠离子电池的循环性能[131]Fig.18 (a) Variations in atomic structure and charge density of Zn-TPyP, Ni-TPyP, Cu-TPyP and Co-TPyP; (b) N 1s XPS spectrum of Zn-TPyP; (c) cyclic performance of sodium-ion batteries constructed with different metal porphyrins. Adapted with permission[131] |
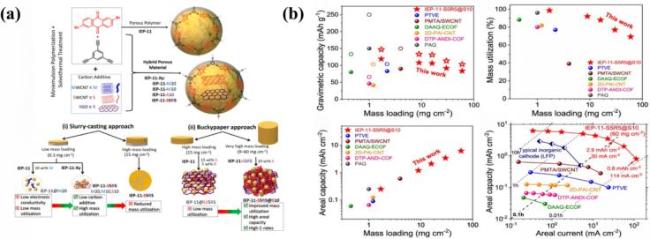
图20 (a) IEP-11聚合物和IEP-11-Xy复合材料的合成图以及电极制备方法; (b) IEP-11-S5R5@S10巴克纸电极在锂离子半电池中的电化学性能与最先进的高质量负载聚合物电极对比, 包括比容量按活性物质质量(空心符号)/总电极质量(实心符号)计算; 电化学活性位点的质量利用率; 面积容量与质量负载的关系; 面积容量与面积电流的关系[139]Fig.20 (a) Diagram of IEP-11 polymer and IEP-11-Xy composite materials, and electrode preparation process; (b) electrochemical performance of IEP-11-S5R5@S10 buckypaper electrodes in lithium-ion half-cells compared to state-of-the-art high-mass-loading polymer electrodes, including specific capacity calculated based on the mass of active material (hollow symbols)/total electrode mass (solid symbols); mass utilization rate of electrochemically active sites; relationship between areal capacity and mass loading; and relationship between areal capacity and current density. Adapted with permission[139] |
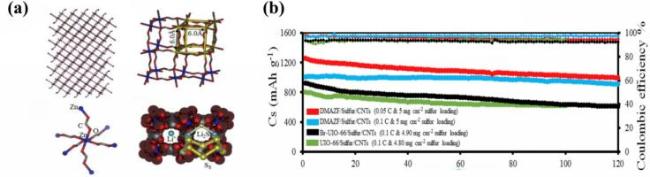
图21 (a) DMAZF晶体结构堆积图、网络窗口尺寸、金属节点以及描述多硫化物抑制过程和锂离子通过纳米孔传输的机制; (b) DMAZF/硫/碳纳米管、Br-UIO-66/硫/碳纳米管和UIO-66/硫/碳纳米管的循环性能[143]Fig.21 (a) DMAZF crystal structure accumulation map, network aperture dimensions, metal nodes and mechanisms describing polysulfide inhibition and Li-ion conduction through nanopores; (b) cyclic properties of DMAZF/Sulfur/CNTs, Br-UIO-66/Sulfur/CNTs, and UIO-66/Sulfur/CNTs. Adapted with permission[143] |
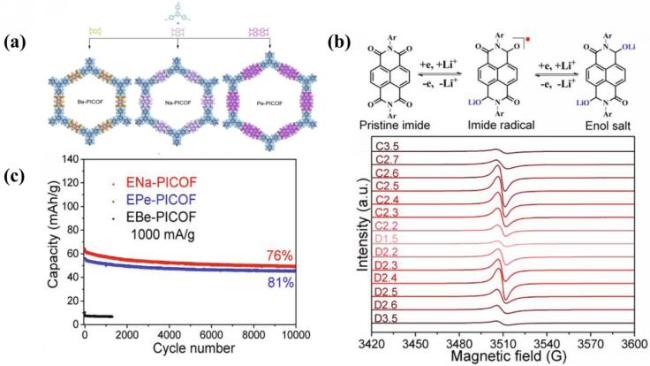
图22 (a) 共价有机框架的化学结构; (b) PICOFs的代表性氧化还原机制以及ENa-PICOF的原位电子顺磁共振光谱; (c) PICOFs在1000 mA·g-1下的循环性能[146]Fig.22 (a) Chemical structure of covalent organic framework; (b) illustrative redox mechanism of PICOFs and in situ electron paramagnetic resonance spectra of ENa-PICOF; (c) cyclic stability of PICOFs at 1000 mA·g-1. Adapted with permission[146] |
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|
| [24] |
|
| [25] |
|
| [26] |
|
| [27] |
|
| [28] |
|
| [29] |
|
| [30] |
|
| [31] |
|
| [32] |
|
| [33] |
|
| [34] |
|
| [35] |
|
| [36] |
|
| [37] |
|
| [38] |
|
| [39] |
|
| [40] |
|
| [41] |
|
| [42] |
|
| [43] |
|
| [44] |
|
| [45] |
|
| [46] |
|
| [47] |
|
| [48] |
|
| [49] |
|
| [50] |
|
| [51] |
|
| [52] |
|
| [53] |
|
| [54] |
|
| [55] |
|
| [56] |
|
| [57] |
|
| [58] |
|
| [59] |
|
| [60] |
|
| [61] |
|
| [62] |
|
| [63] |
|
| [64] |
|
| [65] |
|
| [66] |
|
| [67] |
|
| [68] |
|
| [69] |
|
| [70] |
|
| [71] |
|
| [72] |
|
| [73] |
|
| [74] |
|
| [75] |
|
| [76] |
|
| [77] |
|
| [78] |
|
| [79] |
|
| [80] |
|
| [81] |
|
| [82] |
|
| [83] |
|
| [84] |
|
| [85] |
|
| [86] |
|
| [87] |
|
| [88] |
|
| [89] |
|
| [90] |
|
| [91] |
|
| [92] |
|
| [93] |
|
| [94] |
|
| [95] |
|
| [96] |
|
| [97] |
|
| [98] |
|
| [99] |
|
| [100] |
|
| [101] |
|
| [102] |
|
| [103] |
|
| [104] |
|
| [105] |
|
| [106] |
|
| [107] |
|
| [108] |
|
| [109] |
|
| [110] |
|
| [111] |
|
| [112] |
|
| [113] |
|
| [114] |
|
| [115] |
|
| [116] |
|
| [117] |
|
| [118] |
|
| [119] |
|
| [120] |
|
| [121] |
|
| [122] |
|
| [123] |
|
| [124] |
|
| [125] |
|
| [126] |
|
| [127] |
|
| [128] |
|
| [129] |
|
| [130] |
|
| [131] |
|
| [132] |
|
| [133] |
|
| [134] |
|
| [135] |
|
| [136] |
|
| [137] |
|
| [138] |
|
| [139] |
|
| [140] |
|
| [141] |
|
| [142] |
|
| [143] |
|
| [144] |
|
| [145] |
|
| [146] |
|
| [147] |
|
| [148] |
|
| [149] |
|
| [150] |
|
| [151] |
|
| [152] |
|
| [153] |
|
| [154] |
|
| [155] |
|
| [156] |
|
| [157] |
|
| [158] |
|
/
| 〈 |
|
〉 |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}