
High-Entropy Oxygen Evolution Catalysts: Mechanistic Analysis, Optimization Strategies, and Prospective Challenges
Received date: 2025-07-14
Revised date: 2025-08-09
Online published: 2025-10-25
Supported by
National Natural Science Foundation of China(52072310)
Hydrogen production via water electrolysis powered by renewable energy sources represents a critical approach to addressing the dual challenges of energy and the environment. However, the practical implementationof this technology remains constrained by the sluggish kinetics of the anodic oxygen evolution reaction (OER). Recent advances in high-entropy materials (HEMs) with unique structural configurations and compositional tunability have demonstrated breakthrough capabilities in OER catalysis. Their near-continuous adsorption energy tunability across multi-dimensional landscapes enables surpassing the perforce ceilings of conventional single-/dual-component electrocatalysts. While substantial progress has been achieved in developing HEMs for OER catalysis, formidable scientific challenges persist regarding the intricate composition-structure-activity relationships in multi-component systems and unresolved mechanistic ambiguities governing catalytic synergies. This review systematically examines the fundamental mechanisms underlying the four-electron transfer process in OER, followed by a critical survey of recent breakthroughs in high-entropy alloys (HEAs), high-entropy oxides (HEOs), and high-entropy metal-organic frameworks (HEMOFs) for OER applications. By emphasizing three critical dimensions: atomic coordination environment modulation, electronic structure engineering, and surface adsorption energy optimization, we establish explicit correlations between compositional architecture, structural characteristics, and catalytic performance. This framework profoundly elucidates the synergistic catalytic mechanisms arising from multi-metallic active sites. Furthermore, we propose strategic optimization pathways through material design, defect engineering, and elemental regulation. The review concludes by discussing emerging challenges and future opportunities in this rapidly evolving field. This review can provide inspiration for the accurate design of high-entropy electrocatalysts, the atomic-level analysis of structure-activity relationships, and the regulation and optimization of catalytic performance.
Contents
1 Introduction
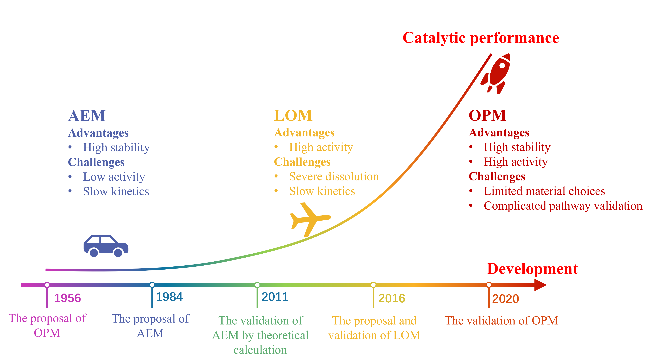
2 OER pathway
2.1 AEM
2.2 LOM
2.3 OPM
3 Research progress and bottlenecks of high‑entropy oxygen evolution catalytic materials
3.1 High‑entropy alloys
3.2 High‑entropy oxides
3.3 High‑entropy MOFs
3.4 Other high‑entropy compounds
4 Optimization strategies
4.1 Machine learning‑assisted design
4.2 Defect engineering
4.3 Element regulation
5 Conclusion and outlook
Shaofu Kuang , Xue Lu , Jianxing Wang , Hua Lin , Qing Li . High-Entropy Oxygen Evolution Catalysts: Mechanistic Analysis, Optimization Strategies, and Prospective Challenges[J]. Progress in Chemistry, 2025 , 37(11) : 1581 -1603 . DOI: 10.7536/PC20250715
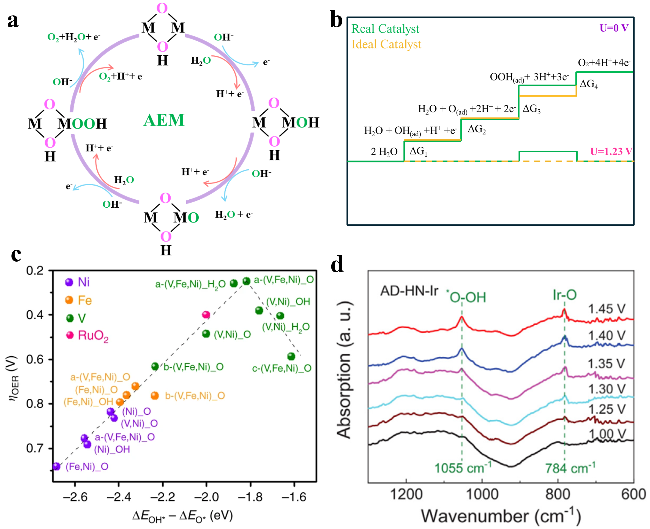
图2 (a) AEM示意图;(b) 实际的和理想催化剂的吉布斯自由能图;(c) 部分氧化物催化剂过电位与ΔGOH*-ΔG*O的火山图[49];(d) AD-NH-Ir在OER过程中的原位红外光谱图[50]Fig.2 (a) Schematic diagram of AEM. (b) The Gibbs free energy diagrams of catalysts under actual and ideal conditions. (c) The volcano relationship between ΔGOH*-ΔG*O and OER overpotential for part oxides[49]. Copyright 2018, Springer Nature. (d) In situ FTIR of AD-NH-Ir during OER[50]. Copyright 2021, Springer Nature |
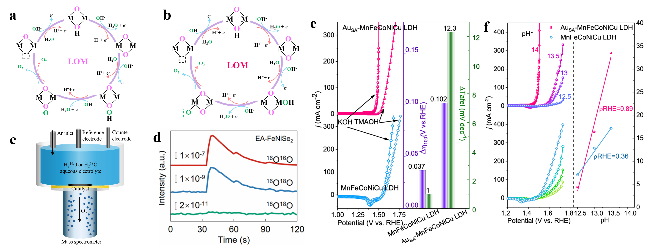
图3 (a,b) LOM及其亚型示意图;(c) 原位差分质谱仪示意图;(d) 18O标记EA-FeNiSe2产生氧气的DEMS测量信号[57];(e) 在1.0 mol/L KOH和1.0 mol/L TMAOH条件下,AuSA-MnFeCoNiCu LDH和MnFeCoNiCu LDH的极化曲线以及在不同电解液中过电位和Tafel斜率的变化(@100 mA·cm-2)[58];(f) AuSA-MnFeCoNiCu LDH和MnFeCoNiCu LDH在不同pH的电解液下的极化曲线(左)以及在1.45 V(相对于可逆氢电极)时电流密度随pH的变化(右)[58]Fig.3 Schematic diagram of (a) LOM and (b) sub-LOM, respectively. (c) Schematic illustration of in situ DEMS device. (d) DEMS signals of O2 products for EA-FeNiSe2 over time[57]. Copyright 2024, American Chemical Society. (e) Polarization curves of AuSA-MnFeCoNiCu LDH and MnFeCoNiCu LDH in 1.0 mol/L KOH and 1.0 mol/L TMAOH, shift of overpotential at 100 mA·cm-2 and Tafel slopes from KOH to TMAOH[58]. Copyright 2023, Springer Nature. (f) Polarization curves measured in KOH electrolytes with different pH (left), current density at 1.45 V (vs. RHE) as a function of pH (right)[58]. Copyright 2023, Springer Nature |
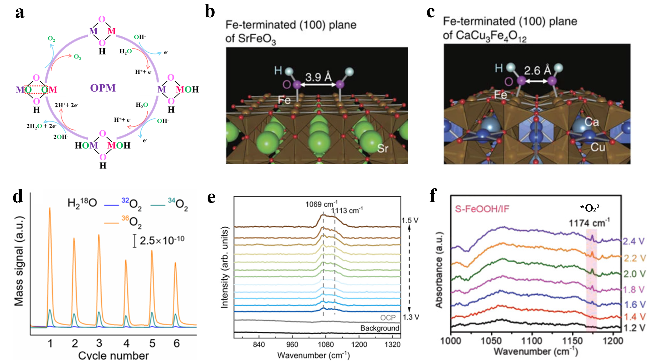
图4 (a) OPM示意图;OH-吸附在(b) SrFeO3和(c) CaCu3Fe4O12晶体结构模型[63];(d) Ru-Co3O4产生氧气的DEMS测量信号[64];(e) Ru/MnO2在OER过程中的原位红外光谱[65];(f) S-FeOOH/IF在OER过程中的原位红外光谱[62]Fig.4 (a) Schematic diagram of OPM. OH- adsorbates on (b) SrFeO3 and (c) CaCu3Fe4O12 crystalline structure models[63]. Copyright 2015, Springer Nature. (d) DEMS signals of O2 products for Ru array-Co3O4[64]. Copyright 2024, American Chemical Society. (e) In situ FTIR of Ru/MnO2 during OER[65]. Copyright 2024, Springer Nature. (f) In situ FTIR of S-FeOOH/IF during OER[62]. Copyright 2022, Wiley |
表1 常温条件下几种典型高熵材料OER的催化活性Table 1 Electrocatalytic activity of some typical high-entropy materials for the OER under ambient temperature conditions |
| Catalyst | Type | Electrolyte | Overpotential (mV) @10 mA·cm-2 | Tafel slope (mV·dec-1) | Stability (V vs. RHE) | Ref |
|---|---|---|---|---|---|---|
| NiCo LDH | binary | 1 mol·L-1 KOH | 271 | 72 | 20 h@1.673 V | 70 |
| Co-P-B | ternary | 1 mol·L-1 NaOH | 290 | 42 | 20 h@1.53 V | 71 |
| Co3V2O8 | ternary | 1 mol·L-1 KOH | 359 | 65 | 3 h@10 mA·cm-2 | 72 |
| FeCo2P | ternary | 0.1 mol·L-1 KOH | 320 | 55 | 12 h@10 mA·cm-2 | 73 |
| S15-CoTe | ternary | 1 mol·L-1 KOH | 255 | 54,7 | 100 h@10 mA·cm-2 | 74 |
| NiCoFeB | quaternary | 1 mol·L-1 KOH | 284 | 46 | 10 h@285 mV | 75 |
| E-FeCoNiZn | quaternary | 1 mol·L-1 KOH | 259 | 37.1 | 48 h@10 mA·cm-2 | 76 |
| AlNiCoIrMo | HEA | 0.5 mol·L-1 H2SO4 | 233 | 55.2 | 48 h@1.52 V | 77 |
| AlCrCuFeNi | HEA | 1 mol·L-1 KOH | 270 | 77.5 | 35 h@290 mV | 78 |
| CoFeGaNiZn | HEA | 1 mol·L-1 KOH | 370 | 71 | 10 h@1.5 V | 79 |
| CoCuFeNiMnMo | HEA | 1 mol·L-1 KOH | 375 | 61 | 72 h@10 mA·cm-2 | 80 |
| PtFeCoNiMn | HEA | 1 mol·L-1 KOH | 357 | 114.6 | 60 h@10 mA·cm-2 | 81 |
| MoZnFeCoNi | HEA | 1 mol·L-1 KOH | 221 | 48.78 | 1550 h@100 mA·cm-2 | 82 |
| CoFeCrMoMnO | HEO | 1 mol·L-1 KOH | 188 | 30 | 24 h@50 mA·cm-2 | 83 |
| ZnFeNiCuCoRu-O | HEO | 1 mol·L-1 KOH | 170 | 56 | 30 h@10 mA·cm-2 | 39 |
| CoCuFeMoOOH | HEO | 1 mol·L-1 KOH | 199 | 48.8 | 72 h@50mA·cm-2 | 84 |
| (FeCoNiCrMn)3O4 | HEO | 1 mol·L-1 KOH | 288 | 60 | 95 h@10 mA·cm-2 | 85 |
| (CoCuFeMnNi)3O4 | HEO | 1 mol·L-1 KOH | 350 | 59.5 | 12 h@15 mA·cm-2 | 86 |
| La0.8Sr0.2Co0.8Fe0.2O3-δ | HEO | 1 mol·L-1 KOH | 248 | 51 | 20 h@15 mA·cm-2 | 87 |
| Ag@CoCuFeAgMoOOH | HEO | 1 mol·L-1 KOH | 218 | 35.3 | 50 h@10 mA·cm-2 | 88 |
| FeCoNiCrVB | HEB | 1 mol·L-1 KOH | 237 | 24.2 | 20 h@1.46 V | 89 |
| FeNiCoCrMnS2 | HES | 1 mol·L-1 KOH | 199 | 39.1 | 55 h@500 mA·cm-2 | 90 |
| FeCoNiPB | HEPB | 1 mol·L-1 KOH | 235 | 53 | 40 h@10 mA·cm-2 | 91 |
| NiCoFeMnCrP | HEP | 1 mol·L-1 KOH | 220 | 94.5 | 24 h@1.55 V | 92 |
| FeCoNiMnCuPx | HEP | 1 mol·L-1 KOH | 239 | 72.5 | 50 h@100 mA·cm-2 | 93 |
| HEMOFs | HEMOF | 1 mol·L-1 KOH | 310 | 48 | 8.3 h@10 mA·cm-2 | 94 |
| FeCoNiCuMn MOF | HEMOF | 1 mol·L-1 KOH | 196 | 55 | 75 @10 mA·cm-2 | 95 |
| MnFeCoNiCu MOF | HEMOF | 1 mol·L-1 KOH | 245 | 54 | 48 h@10 mA·cm-2 | 96 |
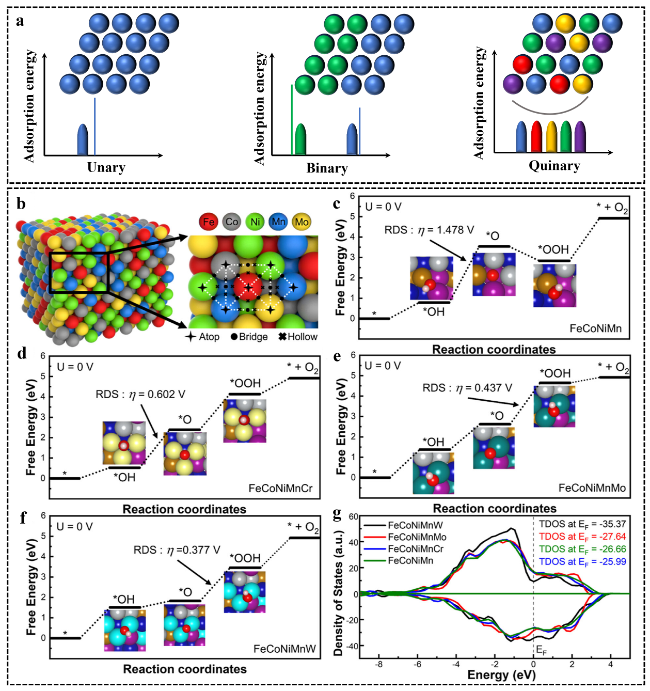
图7 (a) 各类催化剂表面吸附能的示意图;(b) HEAs不同活性位点示意图;(c) FeCoNiMn;(d) FeCoNiMnCr,(e) FeCoNiMnMo和(f) FeCoNiMnW吉布斯自由能变化图;以及HEAs的(g) TDOS图[99]Fig.7 (a) Schematic diagram of the adsorption energy of various catalyst surfaces. (b) Diagrams of different active sites in HEAs. The change in Gibbs free energy of (c) FeCoNiMn, (d) FeCoNiMnCr, (e) FeCoNiMnMo, and (f) FeCoNiMnW. (g) TDOS plots for HEAs[99]. Copyright 2023, American Chemical Society |
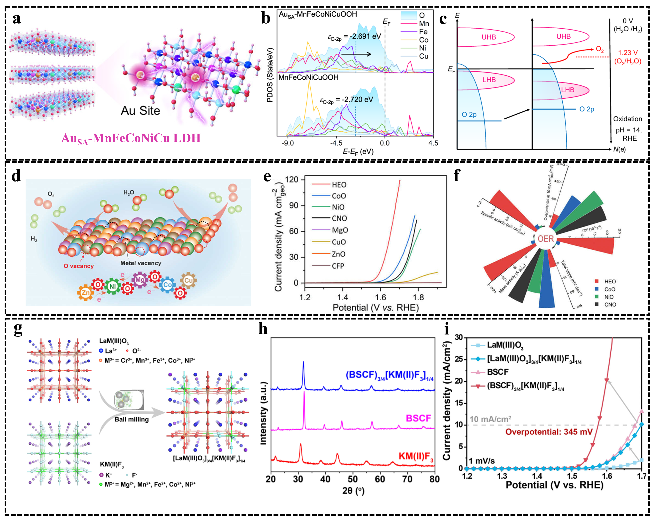
图8 (a) AuSA-MnFeCoNiCu LDH示意图;MnFeCoNiCu/Au-MnFeCoNiCu LDH的(b)态密度和(c)能带示意图[58], (Co0.2Cu0.2Mg0.2Ni0.2Zn0.2)O的(d) OER机理示意图,以及不同氧化物(e) 极化曲线和(f) 多角度对比的玫瑰示意图[113];(g)高熵钙钛矿[LaM(Ⅲ)O3] 3/4[KM(Ⅱ)F3]1/4制备示意图,以及(BSCF)3/4[KM(Ⅱ)F3]1/4的(h) XRD图和(i) 极化曲线[118]Fig.8 (a) Schematic of AuSA-MnFeCoNiCu LDH. (b) Projected density of states of MnFeCoNiCu/Au-MnFeCoNiCu LDH and (c) schematic band diagrams[58]. Copyright 2023, Springer Nature. (d) Schematic diagram of the OER mechanism for (Co0.2Cu0.2Mg0.2Ni0.2Zn0.2)O, and (e) polarization curves and (f) rose diagram showing multi‐angle comparisons[113]. Copyright 2022, Elsevier. (g) Schematic illustration of the synthesis of [LaM(Ⅲ)O3]3/4[KM(Ⅱ)F3]1/4, and (h) XRD image and (i) polarization curves of (BSCF)3/4[KM(Ⅱ)F3]1/4[118]. Copyright 2021, Wiley |
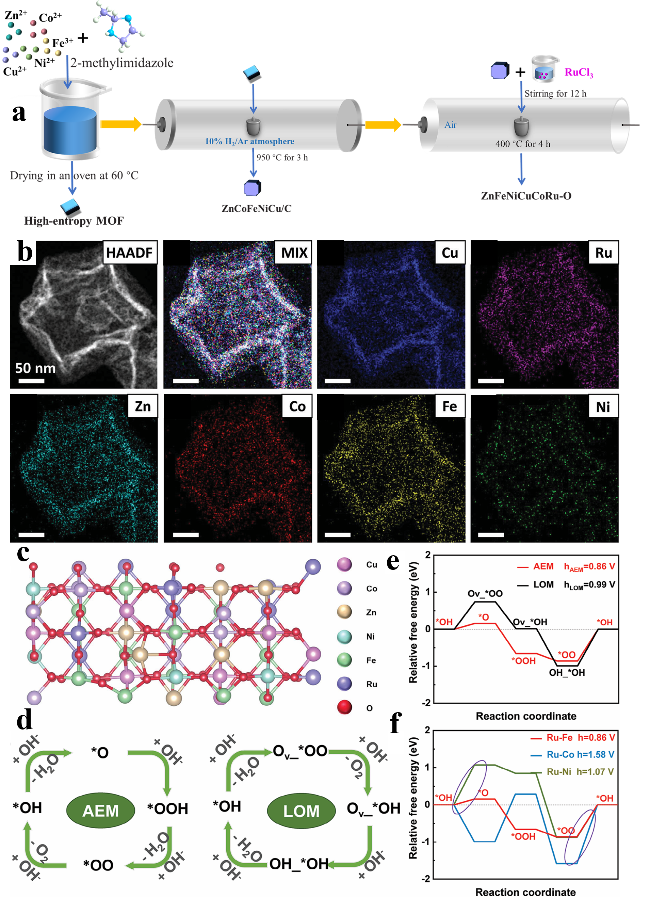
图9 (a) ZnFeNiCuCoRuO制备示意图;(b) HAADF-STEM图像及对应的元素分布图;(c) ZnFeNiCuCoRuO模型;(d) AEM和LOM机理示意图以及其(e) 自由能;(f) 在Ru-Fe、Ru-Co、Ru-Ni位点上AEM的自由能[39]Fig.9 (a) Schematic illustration of the synthesis of ZnFeNiCuCoRuO. (b) HAADF-STEM image and corresponding elemental mapping of ZnFeNiCuCoRuO. (c) The model of ZnFeNiCuCoRuO. (d) Schematic illustration of AEM and LOM. (e) The relative free energy of AEM and LOM pathways. (f) The energetics of AEM on Ru-Fe, Ru-Co, and Ru-Ni site, respectively[39]. Copyright 2023, Wiley |
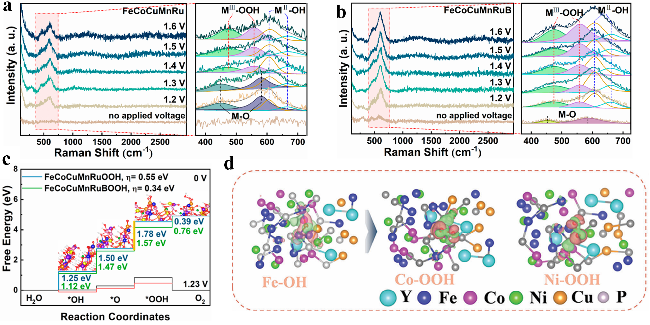
图10 (a) FeCoCuMnRu和(b) FeCoCuMnRuB在不同电压下的原位拉曼[130],以及(c) FeCoCuMnRu和FeCoCuMnRuB吉布斯自由能变化图[130],(d) FeCoNiCuYP/C催化剂中Fe-OH、Co-OOH、Ni-OOH中间体的电荷密度差[135]Fig.10 In situ Raman spectra of (a) FeCoCuMnRu alloy and (b) FeCoCuMnRuB boride at different applied voltages[130]. (c) The change in Gibbs free energy of FeCoCuMnRu alloy and FeCoCuMnRuB boride involved in the OER process[130]. Copyright 2024, American Chemical Society. (d) Differential charge density of Fe-OH, Co-OOH, Ni-OOH intermediate of FeCoNiCuYP/C catalysts[135]. Copyright 2024, Wiley |
| Model | Advantages | Disadvantage |
|---|---|---|
| NN | (1) Powerful for non-linear relationships and complex model | (1) Prone to overfitting with limited training data |
| (2) Robust to noisy data | (2) Requires careful tuning of parameters | |
| (3) Ability to learn from large datasets | (3) Black-box nature makes interpretation difficult | |
| SVM | (1) Effective in high-dimensional feature spaces | (1) Can be slow to train on large datasets |
| (2) Works well with small to medium-sized datasets | (2) Sensitivity to choice of kernel parameters | |
| (3) Versatile due to kernel trick for non-linear classification | (3) Memory-intensive for large-scale problems | |
| RF | (1) High accuracy and robustness | (1) Can be slow to predict on large datasets |
| (2) Effective for high-dimensional feature spaces | (2) Lack of interpretability due to ensemble nature | |
| (3) Handles missing values while preserving accuracy | (3) Potential overfitting on noisy datasets without regularization |
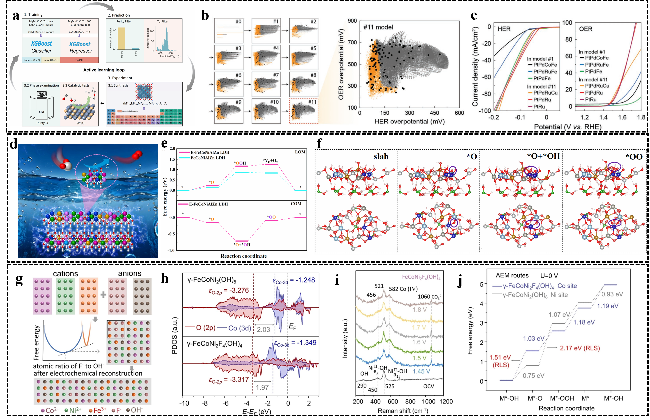
图12 (a) 主动学习过程示意图[146];(b) Pareto主动学习模型在每个步骤的计算结果及对应数据点;(c) 第一个模型和最后一个模型中性能最佳的三种催化剂的HER与OER的极化曲线[147];(d) E-FeCoNiAlZn于缺陷处OER反应机理(LOM亚型-COM)示意图;(e) E-FeCoNiAlZn和FeCoNiAlZn在OER过程中各反应步骤的吉布斯自由能变化图;(f) E-FeCoNiAlZn LDH的COM理论模型涉及*O、*O+*OH和*OO的吸附(黄色、蓝色、白色、灰色、银色、红色、绿色和粉色球分别代表Fe、Co、Ni、Zn、Al、O、B和H)[56];(g) 多种阳离子与阴离子混合反应示意图;(h) γ-FeCoNi2(OH)8和γ-FeCoNi2F4(OH)4中Co与O的态密度图;(i) γ-FeCoNi2F4(OH)4原位拉曼图,(j) γ-FeCoNi2(OH)₈和γ-FeCoNi2F4(OH) 4在AEM路径中吉布斯自由能变化图[160]Fig.12 (a) Training process of AL[146]. Copyright 2024, American Chemical Society. (b) Result of the Pareto active learning model for each step and the corresponding data points. (c) LSV curve of HER and OER for the top three catalysts discovered in the 1st model and the last model[147]. Copyright 2023, Wiley. (d) Schematic illustration of the OER reaction mechanism (sub-LOM, COM) at defect sites in E-FeCoNiAlZn. (e) Free energy for OER of E-FeCoNiAlZn LDH and FeCoNiAlZn LDH. (f) The theoretical models of COM on E-FeCoNiAlZn LDH involved the adsorption of *O, *O+*OH and *OO (The yellow, blue, white, gray, silvery, red, green and pink balls represent Fe, Co, Ni, Zn, Al, O, B and H, respectively)[56]. Copyright 2025, Elsevier. (g) Schematic diagram of the interaction between various cations and anions. (h) Calculated PDOS plots of Co and O for γ-FeCoNi2(OH)8 and γ-FeCoNi2F4(OH)4. (i) In situ Raman spectra of γ-FeCoNi2F4(OH)4. (j) Free energy of γ-FeCoNi2(OH)8 and γ-FeCoNi2F4(OH)4 in the AEM routes[160]. Copyright 2024, Wiley |
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|
| [24] |
|
| [25] |
|
| [26] |
|
| [27] |
|
| [28] |
|
| [29] |
|
| [30] |
|
| [31] |
|
| [32] |
|
| [33] |
|
| [34] |
|
| [35] |
|
| [36] |
|
| [37] |
|
| [38] |
|
| [39] |
|
| [40] |
|
| [41] |
|
| [42] |
|
| [43] |
|
| [44] |
|
| [45] |
|
| [46] |
|
| [47] |
|
| [48] |
|
| [49] |
|
| [50] |
|
| [51] |
|
| [52] |
|
| [53] |
|
| [54] |
|
| [55] |
|
| [56] |
|
| [57] |
|
| [58] |
|
| [59] |
|
| [60] |
|
| [61] |
|
| [62] |
|
| [63] |
|
| [64] |
|
| [65] |
|
| [66] |
|
| [67] |
|
| [68] |
|
| [69] |
|
| [70] |
|
| [71] |
|
| [72] |
|
| [73] |
|
| [74] |
|
| [75] |
|
| [76] |
|
| [77] |
|
| [78] |
|
| [79] |
|
| [80] |
|
| [81] |
|
| [82] |
|
| [83] |
|
| [84] |
|
| [85] |
|
| [86] |
|
| [87] |
|
| [88] |
|
| [89] |
|
| [90] |
|
| [91] |
|
| [92] |
|
| [93] |
|
| [94] |
|
| [95] |
|
| [96] |
|
| [97] |
|
| [98] |
|
| [99] |
|
| [100] |
|
| [101] |
|
| [102] |
(薛世翔, 吴攀, 赵亮, 南艳丽, 雷琬莹. 化学进展, 2022, 34(12): 2686).
|
| [103] |
(周伶俐, 谢瑞刚, 王林江. 化学进展, 2019, 31(2/3): 275).
|
| [104] |
|
| [105] |
|
| [106] |
|
| [107] |
|
| [108] |
|
| [109] |
|
| [110] |
|
| [111] |
|
| [112] |
|
| [113] |
|
| [114] |
|
| [115] |
|
| [116] |
|
| [117] |
|
| [118] |
|
| [119] |
|
| [120] |
|
| [121] |
|
| [122] |
|
| [123] |
|
| [124] |
|
| [125] |
|
| [126] |
|
| [127] |
|
| [128] |
|
| [129] |
|
| [130] |
|
| [131] |
|
| [132] |
|
| [133] |
|
| [134] |
|
| [135] |
|
| [136] |
|
| [137] |
|
| [138] |
|
| [139] |
|
| [140] |
|
| [141] |
|
| [142] |
|
| [143] |
|
| [144] |
|
| [145] |
|
| [146] |
|
| [147] |
|
| [148] |
|
| [149] |
|
| [150] |
|
| [151] |
|
| [152] |
|
| [153] |
|
| [154] |
|
| [155] |
|
| [156] |
|
| [157] |
|
| [158] |
|
| [159] |
|
| [160] |
|
| [161] |
|
| [162] |
|
/
| 〈 |
|
〉 |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}