
Applications of Covalent Organic Frameworks in Electrocatalytic Reduction of CO2
Received date: 2025-04-02
Revised date: 2025-05-13
Online published: 2025-10-25
Supported by
Project of Education Department of Jilin Province(JJKH20240560KJ)
Large emission of carbon dioxide leads to severe global warming effects. Therefore, it is urgent to convert carbon dioxide. Among various transformation technologies, electrocatalytic reduction of CO2 is able to efficiently and continuously convert carbon dioxide. However, the electrocatalytic reduction of CO2 needs to overcome a higher activation barrier. Traditional electrocatalysts such as metals, metal dichalcogenides, transition metal oxides and 2D metal-free catalysts (g-C3N4) are susceptible to inactivation in homogeneous systems and present low electron transfer efficiency, low ability to adsorb and activate carbon dioxide, low reaction kinetics and low selectivity. Covalent organic frameworks (COFs), which are fabricated through covalent bonds, are a class of emerging porous organic polymers. Ordered alignment and π-π interactions between layers facilitate the transportation of charge carriers. High specific surface area and appropriate pore size enable the adsorption of carbon dioxide and generate more active sites as well. All these unique advantages make COFs an ideal candidate for the electrocatalytic reduction of carbon dioxide. In this paper, we first summarize the synthesis and structural diversity of two- and three-dimensional covalent organic frameworks based on topology. Then, the development of 2D and 3D covalent organic frameworks for the electrocatalytic reduction of carbon dioxide is introduced, respectively. Finally, the potential development of COFs for electrochemical carbon dioxide reduction is discussed.
Contents
1 Introduction
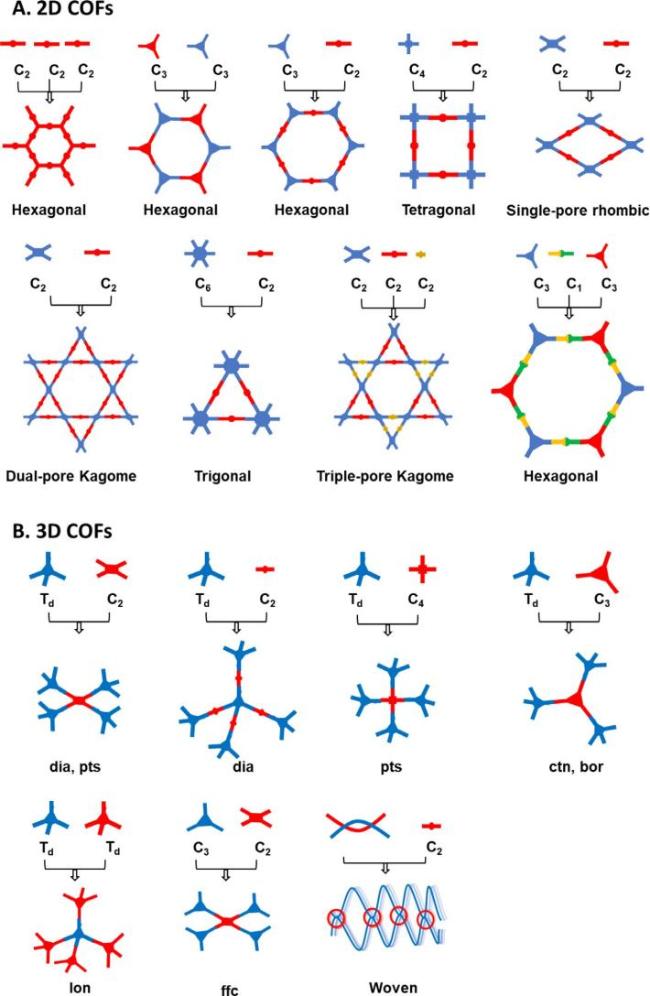
2 Synthesis and structural diversity of COFs
3 COFs for electrocatalytic reduction of carbon dioxide
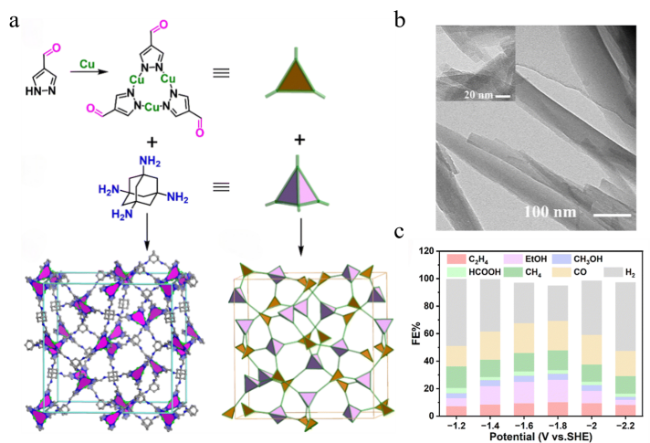
3.1 2D COFs electrocatalysts on CO2 reduction
3.2 3D COFs electrocatalysts on CO2 reduction
4 Conclusion and outlook
Jingyang Li , Dongge Xu , Yunchao Ma , Keyu Cui , Chunbo Liu . Applications of Covalent Organic Frameworks in Electrocatalytic Reduction of CO2[J]. Progress in Chemistry, 2025 , 37(11) : 1622 -1630 . DOI: 10.7536/PC20250401
表1 电催化还原二氧化碳主要产物及还原电势[59]Table 1 Main products of electrocatalytic reduction of CO2 and the reduction potentials[59] |
| Reaction | E0 (V vs. NHE) | Equation |
|---|---|---|
| 2H2O + 2e-→ 2OH-+ H2 | -0.41 | (1) |
| CO2+ 8H++ 8e-→ CH4+ 2H2O | -0.24 | (2) |
| CO2+ 2H++ 2e-→ CO + H2O | -0.51 | (3) |
| CO2+ 6H++ 6e-→ CH3OH + H2O | -0.39 | (4) |
| CO2+ 2H++ 2e-→ HCOOH | -0.58 | (5) |
| 2CO2+ 14H++ 14e-→ C2H6+ 4H2O | -0.27 | (6) |
| 2CO2+ 12H++ 12e-→ C2H5OH + 3H2O | -0.33 | (7) |
| 2CO2+ 2H++ 2e-→ H2C2O4 | -0.87 | (8) |
图3 (a) TT-Por(Co)-COF的合成示意图;(b) TT-Por(Co)- COF与COF-366-Co在不同电势下的FECO;(c) TT-Por(Co)- COF与COF-366-Co在-0.6至-0.9 V的JCO[62]Fig.3 (a) Synthesis of TT-Por(Co)-COF; (b) FECO of TT-Por(Co)-COF and COF-366-Co at different potentials; (c) JCO from -0.6 to -0.9 V of TT-Por(Co)-COF and COF-366-Co Copyright, 2021, Small |
图6 (a) TPE-CoPor-COF,TPTPE-CoPor-COF的合成示意图;(b) TPE-CoPor-COF与TPTPE-CoPor-COF在不同电位势下的一氧化碳法拉第效率;(c) TPE-CoPor-COF与TPTPE-CoPor-COF在不同电位势下的电流密度[64]Fig.6 (a) Synthesis of TPE-CoPor-COF and TPTPE-CoPor-COF; (b) Faradaic efficiency of CO for TPE-CoPor-COF and TPTPE-CoPor-COF at different potentials; (c) current densities of TPE-CoPor-COF and TPTPE-CoPor-COF at different potentials. Copyright 2022, Catalysis Science & Technology |
图7 (a) CoPc-H2Por,CoPc-2H2Por的合成示意图;(b)CoPc-2H2Por一氧化碳和氢气的法拉第效率;(c) CoPc-H2Por一氧化碳和氢气的法拉第效率[65]Fig.7 (a) Synthesis of CoPc-H2Por and CoPc-2H2Por; (b) Faradiac efficiency of CO and H2 for CoPc-2H2Por; (c) Faradiac efficiency of CO and H2 for CoPc-H2Por. Copyright 2022, Advance Materials |
图8 (a) TPPDA-MPor-COFs的合成示意图;(b) TPPDA-MPor-COFs在不同电位势下的法拉第效率;(c) TPPDA-CoPor-COF-NSs在不同电位势下的法拉第效率[66]Fig 8 (a) synthesis of TPPDA-MPor-COFs; (b) FECO of TPPDA-MPor-COFs at different potentials; (c) FECO of TPPDA-CoPor-COF-NSs at different potentials. Copyright 2022, Inorganic Chemistry Frontiers |
图10 (a) MN4-Por、MN3C1-Por、MN2C2-Por的合成示意图;(b) Por-COFs的合成示意图;(c) Co-Por-COFs中CoN4-,CoN3C1-,CoN2C2-为核心的电荷密度差(CDD);(d) 优化的*COOH与(e)*CHOH的吸附构型及电荷密度差[68]Fig. 10 (a) synthesis of MN4-Por, MN3C1-Por, MN2C2-Por; (b) synthesis of Por-COFs; (c) charge density difference (CDD) of CoN4-, CoN3C1-, and CoN2C2- cores of Co-Por-COFs; (d) optimized *COOH and (e) adsorption configurations of *CHOH and charge density difference. Copyright 2023, Small |
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|
| [24] |
|
| [25] |
|
| [26] |
|
| [27] |
|
| [28] |
|
| [29] |
|
| [30] |
|
| [31] |
|
| [32] |
|
| [33] |
|
| [34] |
|
| [35] |
|
| [36] |
|
| [37] |
|
| [38] |
|
| [39] |
|
| [40] |
|
| [41] |
|
| [42] |
|
| [43] |
|
| [44] |
|
| [45] |
|
| [46] |
|
| [47] |
|
| [48] |
|
| [49] |
|
| [50] |
|
| [51] |
|
| [52] |
|
| [53] |
|
| [54] |
|
| [55] |
|
| [56] |
|
| [57] |
|
| [58] |
|
| [59] |
|
| [60] |
|
| [61] |
|
| [62] |
|
| [63] |
|
| [64] |
|
| [65] |
|
| [66] |
|
| [67] |
|
| [68] |
|
| [69] |
|
| [70] |
|
| [71] |
|
/
| 〈 |
|
〉 |










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}