
Preparation of Ionogels and Strategies for Strengthening Its Mechanical Properties
Received date: 2025-05-06
Revised date: 2025-07-13
Online published: 2025-10-30
Supported by
National Natural Science Foundation of China(22378253)
China Postdoctoral Science Foundation(2024M761895)
Shaanxi Provincial Department of Education Science Research Program(24JK0357)
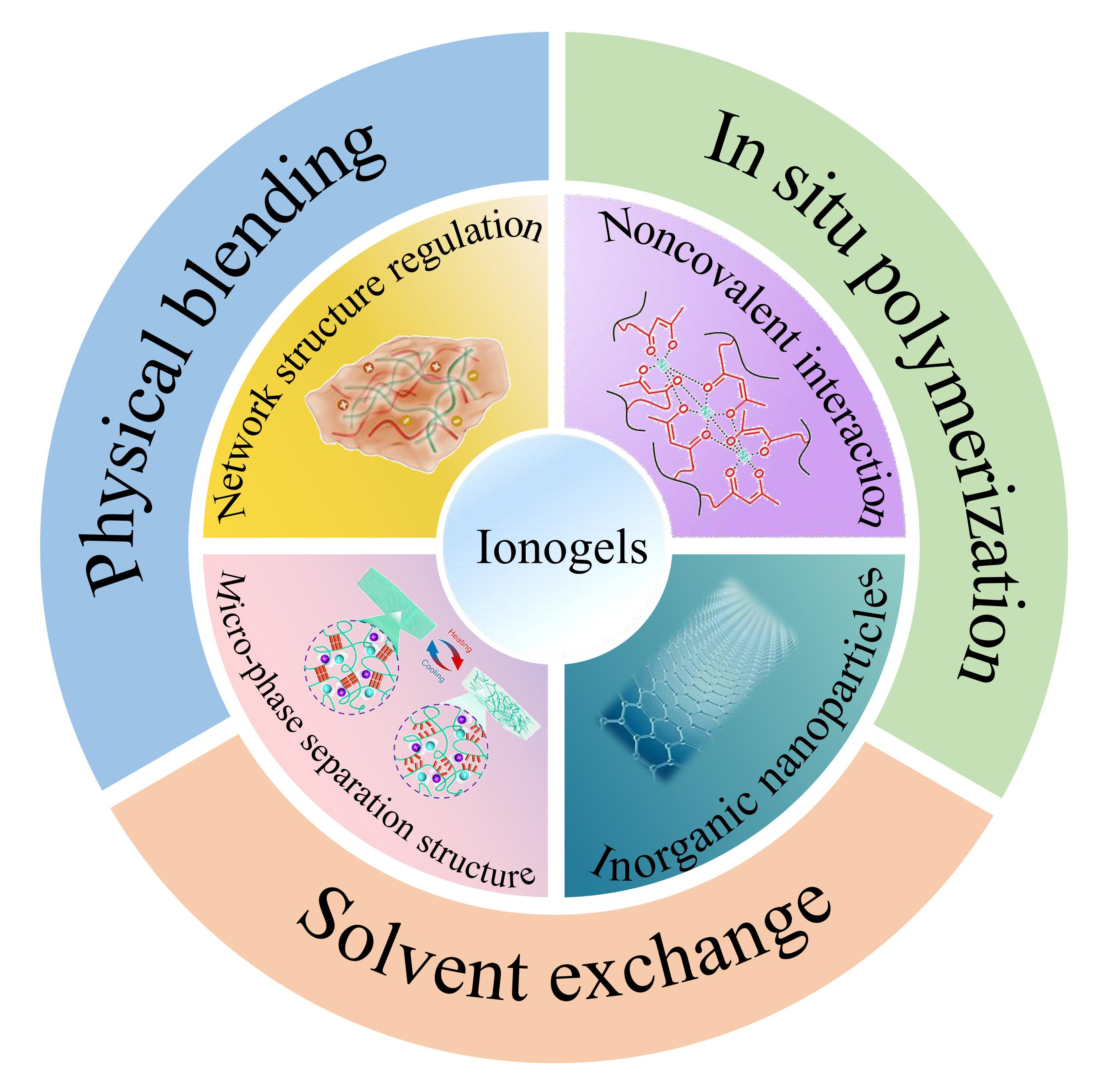
In recent years, flexible electronic devices have shown broad application prospects in fields such as smart sensing equipment, human-machine interfaces and bio-inspired electronic skins. Ionogels demonstrate significant potential in the preparation of flexible electronics due to their excellent electrochemical performance, tunable mechanical properties and high environmental adaptability. However, the generally poor mechanical properties of ionogels limit their widespread use. To address this, this article systematically reviews the research progress of ionogels from two aspects: preparation methods and mechanical reinforcement strategies. First common types of ionic liquids and their characteristics are summarized based on the types of anions and cations. Then the preparation techniques for ionogels are categorized into physical blending, in situ polymerization and solvent exchange, with detailed analysis of their advantages and disadvantages. Next, representative strategies for enhancing mechanical performance are outlined, including regulating polymer network structures, constructing non-covalent interactions, forming microphase-separated structures and introducing inorganic nanoparticles. The mechanism of these strategies, the regulatory effect on the mechanical properties of ionogels, and the application scenarios are systematically explained. Finally, key challenges in current ionogels preparation processes are discussed along with future development directions. This work provides a theoretical foundation for designing high-performance ionogels and improving their properties.
Contents
1 Introduction
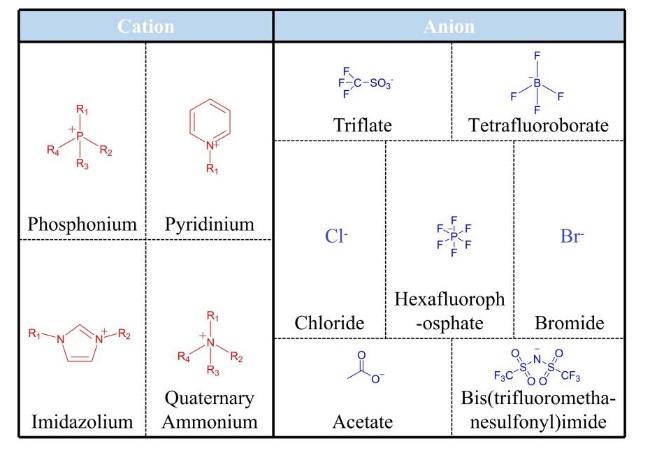
2 Types and characteristics of ionic liquids
3 Preparation methods of ionogels
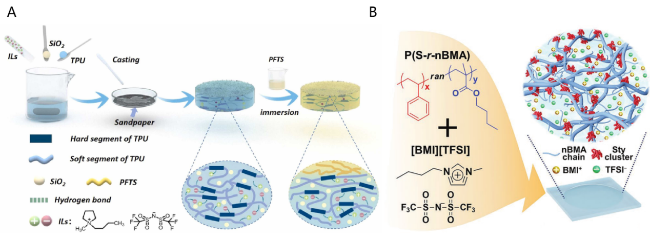
3.1 Physical blending method
3.2 In situ polymerization
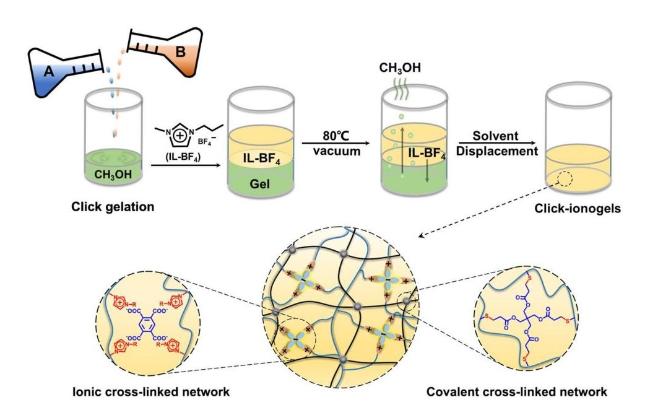
3.3 Solvent exchange
4 Strategies for strengthening the mechanical properties of ionogels
5 Conclusion and outlook
Yan Bao , Junbin Zhou , Ruyue Guo . Preparation of Ionogels and Strategies for Strengthening Its Mechanical Properties[J]. Progress in Chemistry, 2025 , 37(11) : 1674 -1687 . DOI: 10.7536/PC20250501
表1 各类典型ILs的性能对比Table 1 Performance comparison of various typical ILs |
| Types | Representative system | Key characteristics | Applicable scenarios | Biological environmental toxicity |
|---|---|---|---|---|
| Imidazolium | [EMIM][TFSI] | High conductivity, wide electrochemical window, high-temperature resistance (300 ℃), low volatility | Flexible sensors, energy storage devices, electrolytes | Medium-high cytotoxicity, high environmental risk |
| Quaternary Ammonium | [TBA][Ac] | High biocompatibility, biodegradability, low toxicity, thermal stability (>200 ℃) | Bioelectronics, medical sensing, drug carriers | Low-medium cytotoxicity, low-medium environmental risk |
| Phosphonium | [P6,6,6,14][PF6] | Extreme high-temperature resistance (410 ℃), strong hydrophobicity, low viscosity, chemical inertness | High-temperature lubrication, encapsulation materials for extreme environments | High cytotoxicity, high environmental risk |
| Pyridinium | [C5H5NH][BF4] | Adjustable acidity, high catalytic activity, strong dissolving capability | Nitration reaction catalysts, energetic materials synthesis | High cytotoxicity, high environmental risk |
表2 离子凝胶制备策略的优缺点分析及适用体系Table 2 Advantages and disadvantages of ionogel preparation strategy and its applicable systems |
| Methods | Principle | Advantages | Disadvantages | Applicable systems |
|---|---|---|---|---|
| Physical Blending | Mechanically mix polymers with ILs for rapid shaping | Simple process flow, low cost, suitable for rapid preparation of ionogels | Poor compatibility between polymers and ILs, prone to phase separation | Polarity matched polymer-ILs systems, or composite systems containing compatibilizers (such as surfactants) |
| In situ Polymerization | Use ILs as reaction media to directly construct cross-linked networks during monomer polymerization | Precise control of networks structures; strong interfacial bonding between ILs and polymers | High viscosity or chemical inhibition of ILs may lead to uneven polymerization; harsh reaction conditions | Systems with low-viscosity ILs and monomers capable of free-radical polymerization, or systems requiring in situ construction of multi/interpenetrating networks |
| Solvent Exchange | Achieve uniform ILs distribution via solvent exchange | Overcomes solubility limitations; avoids compatibility issues from physical blending | Complex process flow, long preparation time; risk of toxic solvent residues | Systems combining poorly soluble polymers with low-volatility ILs, requiring stepwise solvent exchange for uniform dispersion |
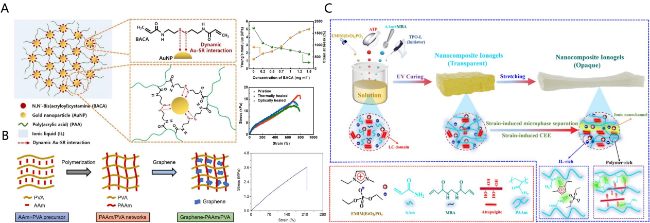
图9 (A) 自修复离子凝胶的结构及其力学性能示意图[77];(B) Gr-PAM/PVA离子凝胶的合成过程及力学性能示意图[78];(C) 纳米复合各向异性离子凝胶的制备示意图[81]Fig.9 (A) Structure and mechanical properties of self-healing ionic gel[77]; (B) synthesis process and mechanical properties of Gr-PAM/PVA ionogel[78]; (C) preparation of nanocomposite anisotropic ionogel[81] |
表3 离子凝胶的力学性能强化策略Table 3 Strategies for strengthening the mechanical properties of ionogel |
| Strategy category | Specific methods | Energy Dissipation mechanism | Advantages | Limitations | Applicable scenarios |
|---|---|---|---|---|---|
| Regulating polymer network structures | Multi-network: Synergistic design of rigid, flexible, and dynamic networks | Fracture of rigid networks, chain slippage of flexible networks, dynamic networks reorganization | Multi-level energy dissipation, high stretchability, toughness | Irreversible fracture of rigid networks, poor compatibility between networks, complex crosslinking process | Joint structure, wearable protective equipment or bionic load-bearing materials of soft robot, etc |
| IPN/SIPN: Interpenetrating crosslinked networks and linear chain segments | Interfacial slippage, inter-phase stress transfer, physical entanglement | Wide-temperature stability, non-bonded interlocking mechanism | Poor mechanical stability, limited dynamic response | Biomedical materials, etc | |
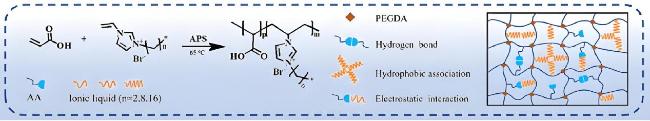
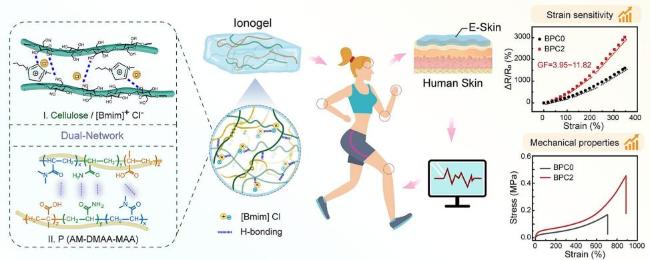
| Constructing non-covalent interactions | Electrostatic interactions, dipole-dipole interactions, ionic bonds, hydrogen bonds | Dynamic bonds breakage and reorganization, reversible rupture of physical/chemical crosslinks | Selective mechanical enhancement, no additional treatment required | Complex preparation, reliance on ligand designs, difficulty in crosslink density control, poor long-term stability | Wearable electronic devices, skin mounted flexible biosensors, and bionic soft materials requiring adaptive functions |
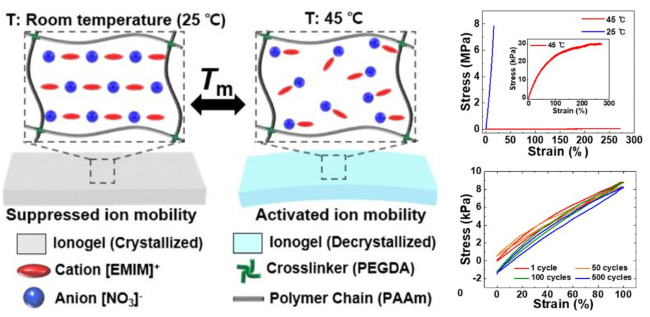
| Forming microphase-separated structures | Introducing phase-transition factors | Phase transition-induced chains rearrangement, crystalline-amorphous transition | Dynamic smart response, wide-temperature adaptability | Limited phase transition temperature, poor cyclic thermal stability | Solid electrolyte field, microfluidic chip, etc |
| Designing phase-separated structures | Fracture of non-covalent bonds in the rigid phase and chains slippage in the flexible phase | Ultra-high strength and toughness, optimized performance via bicontinuous structure | Complex compatibility designs | ||
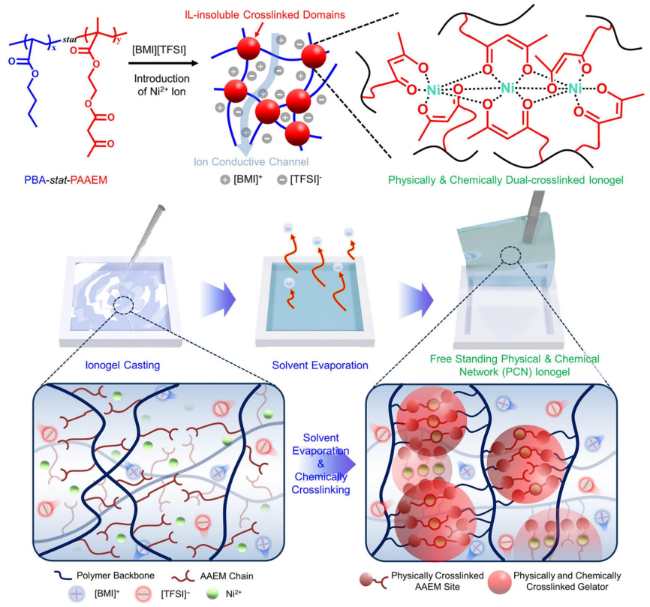
| Introducing inorganic nanoparticles | Crack bridging | INPs bridging crack sides to hinder propagation, enhanced fracture energy via interfacial bonding | Significant crack resistance, simple physical doping, multi-mechanism synergy | INPs aggregation, strict dispersion uniformity, precise filler content control, high costs, complex designs | Sensing under extreme conditions such as automotive electronics, aerospace, etc |
| Energy dissipation | Interface friction, plastic flow, nano-pullout effects | ||||
| Networks regulation | INPs-induced increase in crosslinking density, strain-induced phase separation |
| [1] |
(董昊, 范雨薪, 张旭, 韦会鸽, 徐一飞, 曾威. 复合材料学报, 2025, 42(01): 104).
|
| [2] |
|
| [3] |
(彭玉鑫, 陈雪垠, 章阳坤. 材料工程, 2024, 52(08): 42).
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
(赵文超. 华中科技大学博士论文, 2024).
|
| [12] |
|
| [13] |
(铁建飞. 东华大学博士论文, 2024).
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
(上官小东. 西北大学博士论文, 2009).
|
| [19] |
(金艳, 张桂荣, 窦茜茜, 陈建英, 边玲, 李大伟, 贾庆文. 食品与药品, 2020, 22(3): 242).
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|
| [24] |
|
| [25] |
|
| [26] |
|
| [27] |
|
| [28] |
|
| [29] |
|
| [30] |
|
| [31] |
|
| [32] |
|
| [33] |
|
| [34] |
|
| [35] |
|
| [36] |
|
| [37] |
|
| [38] |
|
| [39] |
|
| [40] |
|
| [41] |
|
| [42] |
|
| [43] |
|
| [44] |
|
| [45] |
|
| [46] |
|
| [47] |
|
| [48] |
|
| [49] |
|
| [50] |
|
| [51] |
|
| [52] |
|
| [53] |
|
| [54] |
|
| [55] |
|
| [56] |
|
| [57] |
|
| [58] |
|
| [59] |
|
| [60] |
|
| [61] |
|
| [62] |
|
| [63] |
|
| [64] |
|
| [65] |
|
| [66] |
|
| [67] |
|
| [68] |
|
| [69] |
|
| [70] |
|
| [71] |
|
| [72] |
|
| [73] |
|
| [74] |
|
| [75] |
|
| [76] |
|
| [77] |
|
| [78] |
|
| [79] |
|
| [80] |
|
| [81] |
|
/
| 〈 |
|
〉 |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}