
Hydrogen Production via Seawater Electrolysis: Current Advances and Future Perspectives on Process Optimization and System Integration
Received date: 2025-05-22
Revised date: 2025-07-04
Online published: 2025-12-10
Supported by
Key Research and Development Program of Heilongjiang Province(2024ZXJ03C06)
Natural Science Foundation of Heilongjiang Province(YQ2022E027)
National Natural Science Foundation of China(52476192)
As a clean and efficient secondary energy source, hydrogen energy represents a strategic pillar for future energy transition, capable of replacing fossil fuels to achieve deep decarbonization in industries, transportation, and other sectors. In recent years, seawater electrolysis has emerged as a promising route for green hydrogen production, owing to its potential to utilize seawater as a feedstock and address offshore wind power utilization challenges in remote marine areas. However, current research on seawater electrolysis predominantly focuses on catalyst development at the material level, with insufficient attention to synergistic optimization at the system and process levels. To bridge this gap, this review systematically summarizes the state-of-the-art technologies and future trends in seawater electrolysis systems and processes. The system is decomposed into four key components: electrolyzer, power supply system, gas-liquid separation system, and gas purification system, with a comprehensive analysis of their current research progress. Additionally, this paper highlights innovations in non-catalyst aspects, including technological and methodological advancements. Finally, future directions and application prospects for seawater electrolysis systems are discussed, emphasizing the importance of integrated system design, scalability, and cost-effectiveness to accelerate industrial deployment. This work aims to provide insights into the holistic development of seawater electrolysis technology for sustainable hydrogen production.
1 Introduction
2 Principles, types of technologies and challenges of hydrogen production by electrolysis of water and seawater
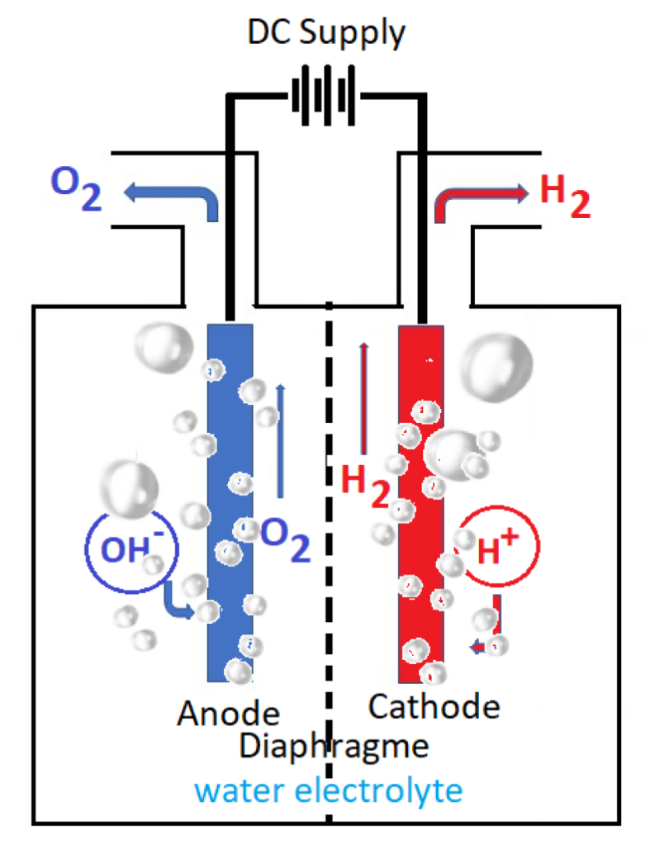
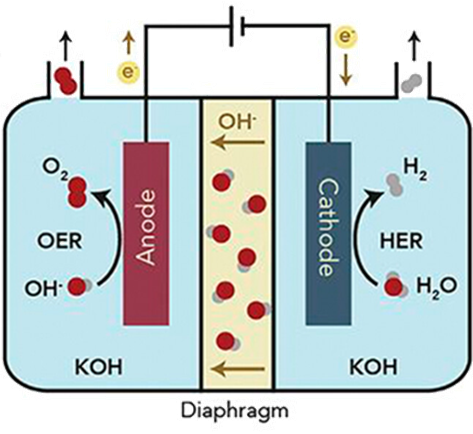
2.1 Hydrogen production by water electrolysis
2.2 Hydrogen production by sea water electrolysis
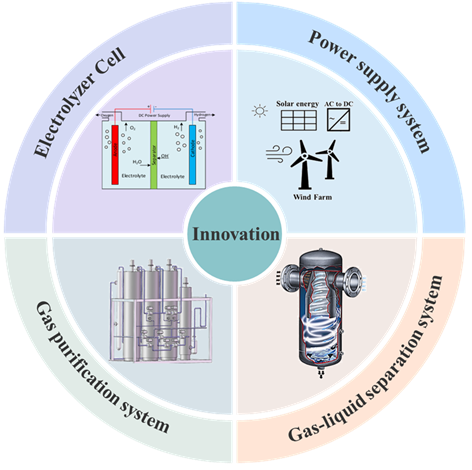
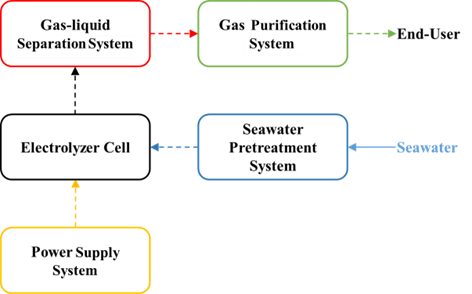
3 Hydrogen production system and process by seawater electrolysis
3.1 Seawater pretreatment system
3.2 Electrolyzer cell
3.3 Power supply system
3.4 Gas-liquid separation system
3.5 Gas purification system
4 Innovation in the process of hydrogen production by seawater electrolysis
4.1 Innovation of the electrolyzer
4.2 Innovation in water electrolysis method
5 Conclusions and prospects
Dongyi Liu , Miaoting Sun , Yang Yu , Jiaxiang Chen , Yanting Zhou , Xingxing Wang , Wei Zhou . Hydrogen Production via Seawater Electrolysis: Current Advances and Future Perspectives on Process Optimization and System Integration[J]. Progress in Chemistry, 2025 , 37(12) : 1877 -1901 . DOI: 10.7536/PC20250519
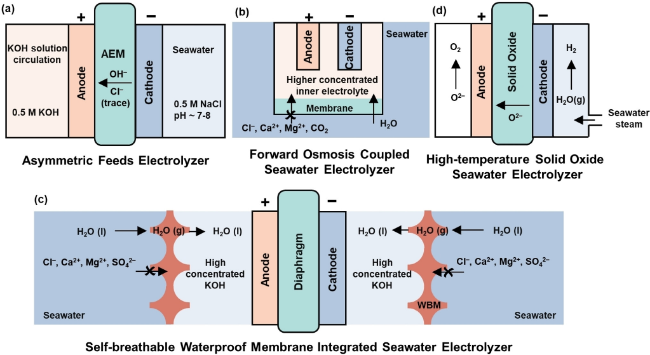
图9 膜电解槽的四大创新方向:(a) 不对称进料电解槽;(b) 正渗透耦合海水电解槽; (c) 自透气防水膜集成海水电解槽;(d) 高温固体氧化物海水电解槽[119]Fig.9 Four major innovations of membrane electrolyzers: (a) asymmetric feed electrolyzer; (b) positive osmosis coupling seawater electrolyzer; (c) self-breathable waterproof membrane integrated seawater electrolyzer; (d) high-temperature solid oxide seawater electrolyzer[119] |
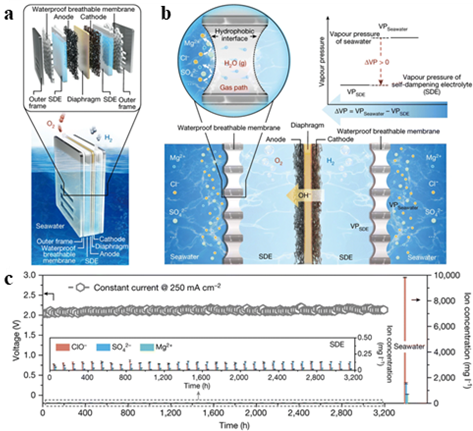
图12 (a) 新型海水电解槽示意图;(b) 通过液-气-液相变的水净化和迁移机制;(c) 在 250 mA·cm-2的恒定电流密度下对放大电解槽的电解耐久性测试[125]Fig.12 (a) Schematic diagram of a new seawater electrolyzer. (b) Water purification and migration mechanism through liquid-gas-liquid phase transition. (c) Electrolytic durability test of an amplified electrolyzer at a constant current density of 250 mA·cm-2[125] |
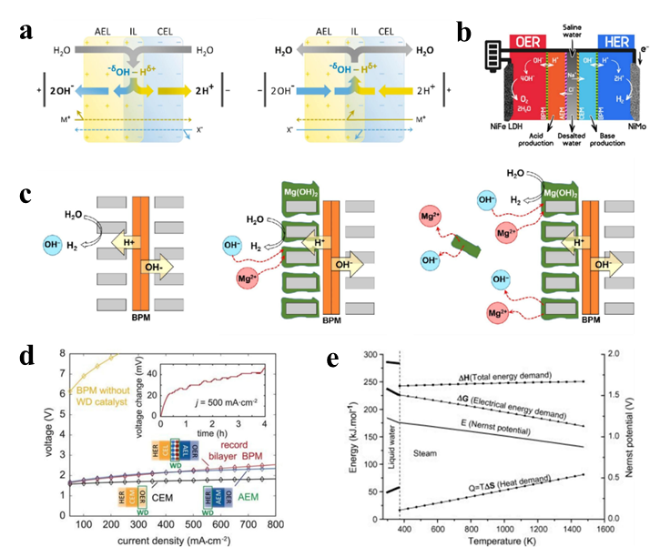
图13 (a) 双极膜原理的示意图[129];(b) 带有AEM、CEM和BPM的海水淡化耦合电催化装置[130];(c) BPM中的无机沉淀和质子通量会以阴极的形式酸化海水[128]Fig.13 (a) Schematic diagram of the bipolar membrane principle[129]. (b) Coupled electrocatalytic device for desalination with AEM, CEM and BPM [130]. (c) Inorganic precipitation and proton fluxes in BPM acidify seawater in the form of cathodes [128] |
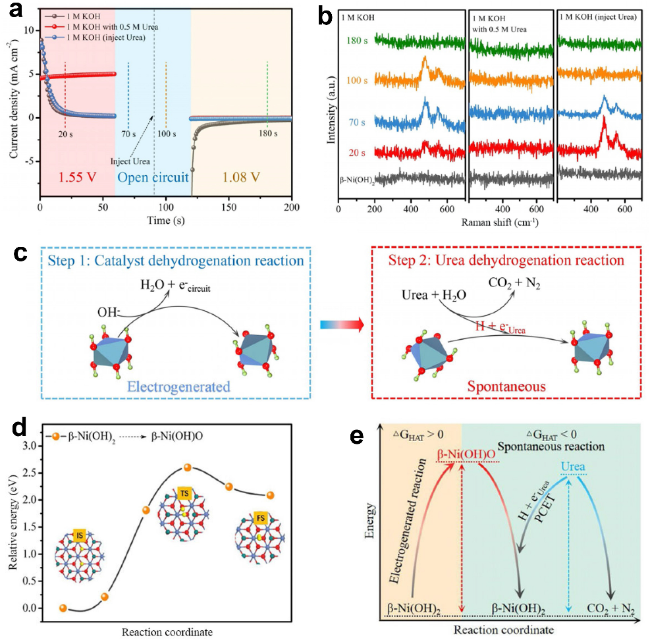
图14 (a) β-Ni(OH)2催化剂在不同电位下的UOR的周期性计时安培测试。 (b) β-Ni(OH)2电极的原位拉曼光谱。(c) β-Ni(OH)2电极上的UOR机制。(d) DFT计算的反应中间体的相对能量。(e) UOR机制的图示[138]Fig.14 (a) Periodic chronometric amperometric testing of UOR of β-Ni(OH)2 catalyst at different potentials. (b) In-situ Raman spectroscopy of the β-Ni(OH)2 electrode. (c) UOR mechanism on the β-Ni(OH)2 electrode. (d) DFT-calculated relative energy of the reaction intermediate. (e) Diagram of the UOR mechanism[138] |
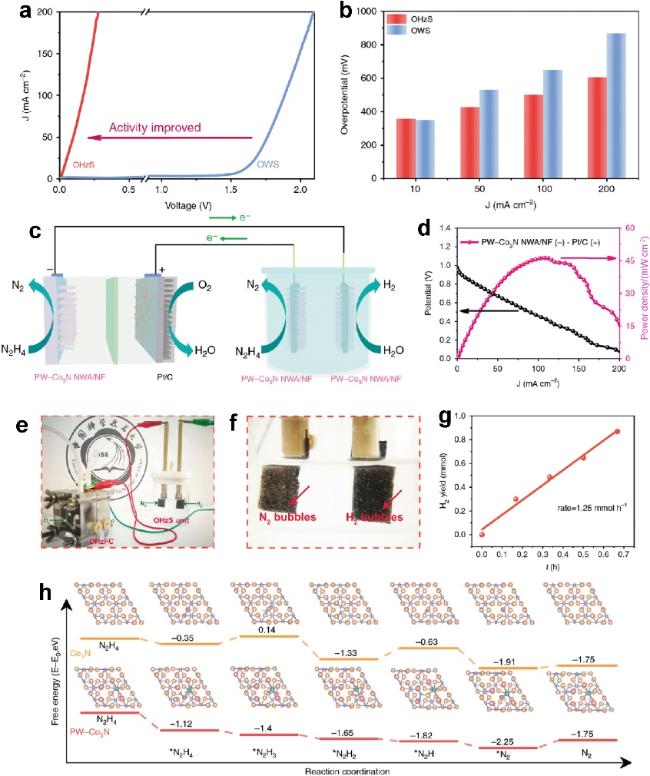
图15 PW-Co3N NWA/NF的电化学性能测量。(a) OHzS和OWS在含有0.1 M肼的1.0 M KOH中电极上的LSV曲线。(b) OHzS和OWSP在不同电流密度下的过电位比较。(c) 将DHzFC和OHzS耦合以实现自供电H2生产。(d) 用于DHzFC的Pt/C电极PW-Co3N NWA/NF的功率密度(P)和电压(V)-电流密度(J)曲线。(e) 自供电H2发生系统的照片。(f) H2生成系统产生的气泡照片。(g) H2代系统不同时间的H2产率[142]Fig.15 Electrochemical performance measurements of PW-Co3N NWA/NF. (a) LSV curves on the OHzS and OWS electrodes in a 1.0 M KOH containing 0.1 M hydrazine. (b) Overpotential comparison of OHzS and OWSP at different current densities. (c) Coupling DHzFC and OHzS for self-powered H2 production. (d) Power density (P) and voltage (V)-current density (J) curves of the Pt/C electrode PW-Co3N NWA/NF for DHzFC. (e) Photograph of the self-powered H2 generation system. (f) Photographs of bubbles generated by the H2 generation system (g) H2 yield of the H2 generation system at different times [142] |
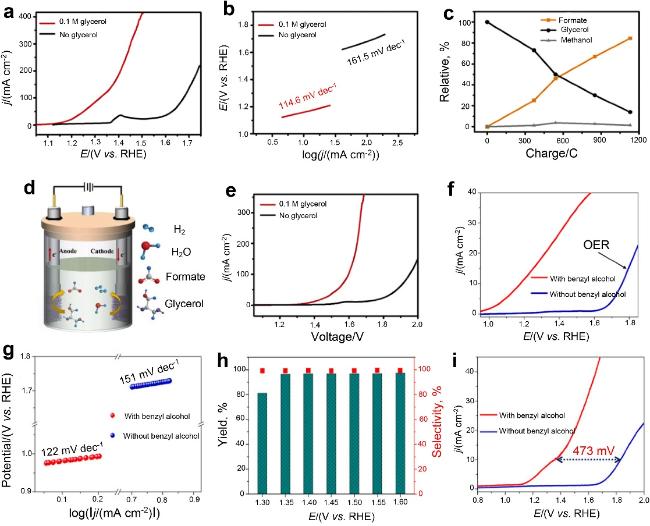
图16 NC/Ni-Mo-N/NF 的电化学性能测量。(a) 含有0.1 M甘油的1 M KOH中GOR的LSV曲线。(b) 相应的塔菲尔斜率。(c) 甘油的消耗量以及甲酸盐和甲醇的收率。(d) HER与GOR整合的示例。(e) 混合电解槽上甘油电化学氧化的LSV曲线[143]。(f) 苯甲醇电化学氧化对Ru-NPs@NCNTs的LSV曲线。(g) 相应的塔菲尔斜率。(h) 苯甲醛在各种电位下的选择性和产率。(i) 苯甲醇在Pt/C||Ru-NPs@NCNTs电解槽[144]Fig.16 Electrochemical performance measurements of NC/Ni-Mo-N/NF. (a) LSV curve of GOR in 1 M KOH with 0.1 M glycerol. (b) Corresponding tafel slope. (c) Consumption of glycerol and yield of formate and methanol. (d) Example of integration of HER with GOR. (e) LSV curve of electrochemical oxidation of glycerol on a hybrid electrolyzer[143]. (f) LSV curves of benzyl alcohol electrochemical oxidation of Ru-NPs@NCNTs. (g) Corresponding tafel slope. (h) Selectivity and yield of benzaldehyde at various potentials. (i) Benzyl alcohol at Pt/C||RU-NPs@NCNTs Electrolyzer[144] |
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|
| [24] |
|
| [25] |
|
| [26] |
|
| [27] |
|
| [28] |
|
| [29] |
|
| [30] |
|
| [31] |
|
| [32] |
|
| [33] |
|
| [34] |
|
| [35] |
|
| [36] |
|
| [37] |
|
| [38] |
|
| [39] |
|
| [40] |
|
| [41] |
|
| [42] |
|
| [43] |
|
| [44] |
|
| [45] |
|
| [46] |
|
| [47] |
|
| [48] |
|
| [49] |
|
| [50] |
|
| [51] |
|
| [52] |
|
| [53] |
|
| [54] |
|
| [55] |
|
| [56] |
|
| [57] |
|
| [58] |
|
| [59] |
|
| [60] |
|
| [61] |
|
| [62] |
|
| [63] |
|
| [64] |
|
| [65] |
|
| [66] |
|
| [67] |
|
| [68] |
|
| [69] |
|
| [70] |
|
| [71] |
|
| [72] |
|
| [73] |
|
| [74] |
|
| [75] |
|
| [76] |
|
| [77] |
|
| [78] |
|
| [79] |
|
| [80] |
|
| [81] |
|
| [82] |
|
| [83] |
|
| [84] |
Purnami,
|
| [85] |
|
| [86] |
|
| [87] |
|
| [88] |
|
| [89] |
|
| [90] |
|
| [91] |
|
| [92] |
|
| [93] |
|
| [94] |
|
| [95] |
|
| [96] |
|
| [97] |
|
| [98] |
|
| [99] |
|
| [100] |
|
| [101] |
|
| [102] |
|
| [103] |
|
| [104] |
|
| [105] |
|
| [106] |
|
| [107] |
|
| [108] |
|
| [109] |
|
| [110] |
|
| [111] |
|
| [112] |
|
| [113] |
|
| [114] |
|
| [115] |
|
| [116] |
|
| [117] |
|
| [118] |
|
| [119] |
|
| [120] |
|
| [121] |
|
| [122] |
|
| [123] |
|
| [124] |
|
| [125] |
|
| [126] |
|
| [127] |
|
| [128] |
|
| [129] |
|
| [130] |
|
| [131] |
|
| [132] |
|
| [133] |
|
| [134] |
|
| [135] |
|
| [136] |
|
| [137] |
|
| [138] |
|
| [139] |
|
| [140] |
|
| [141] |
|
| [142] |
|
| [143] |
|
| [144] |
|
/
| 〈 |
|
〉 |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}