
Research Progress on Surface Reconstruction Regulated Oxygen Evolution Electrocatalyst Performance
Received date: 2025-06-10
Revised date: 2025-10-16
Online published: 2026-02-03
Supported by
National Natural Science Foundation of China(52472242)
During the oxygen evolution reaction (OER),the surface reconstruction phenomenon of catalysts is closely related to the enhancement of their catalytic performance. However,the mechanistic understanding of catalyst surface reconstruction remains incomplete,particularly the technical bottlenecks in achieving controlled surface reconstruction and precise regulation of active sites. To address this,this article systematically elucidates two OER catalytic mechanisms-the adsorbate evolution mechanism (AEM) and the lattice oxygen oxidation mechanism (LOM) and analyzes the influence of pH,temperature,and applied potential on the surface reconstruction behavior of catalysts. Key mechanisms such as ion leaching (cation/anion leaching),elemental doping (metal/non-metal doping),and size effect modulation are summarized to reveal the relationship between surface reconstruction and catalytic activity of the OER catalysts. This work aims to provide theoretical support for the development of high-performance OER electrocatalysts. Finally,based on the challenges and prospects faced by surface-reconstructed OER catalysts,the potential impact of controlled reconstruction on the catalytic performance is prospected.
Contents
1 Introduction
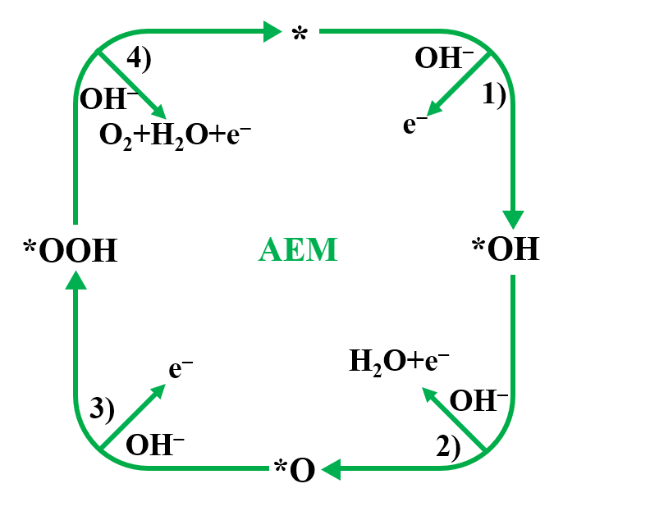
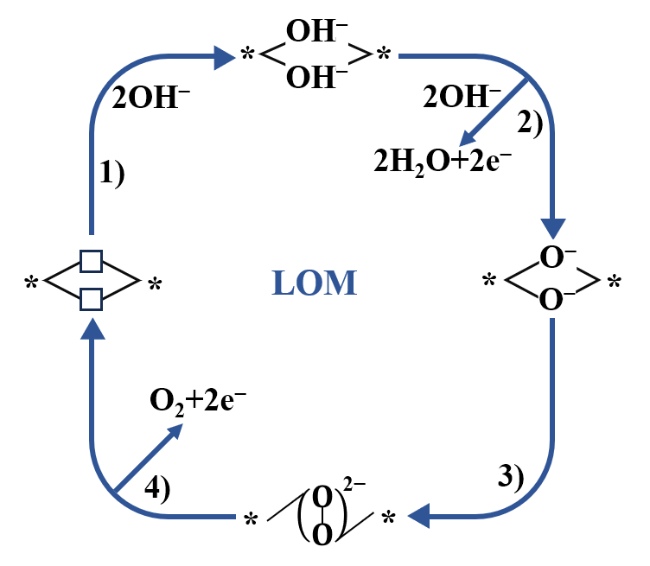
2 OER catalytic mechanisms
2.1 Adsorbate evolution mechanism
2.2 Lattice oxygen oxidation mechanism
3 Surface reconstruction
3.1 Fundamental principles of surface reconstruction
3.2 Factors influencing surface reconstruction
4 Strategies for modulating oer catalyst surface reconstruction
4.1 Ion leaching
4.2 Elemental doping
4.3 Size regulation
5 Conclusion and outlook
Junjie Wen , Lixiang Ding , Zhen Yuan , Junyi Zhang , Wen Lei , Haijun Zhang . Research Progress on Surface Reconstruction Regulated Oxygen Evolution Electrocatalyst Performance[J]. Progress in Chemistry, 2026 , 38(2) : 237 -251 . DOI: 10.7536/PC20250609
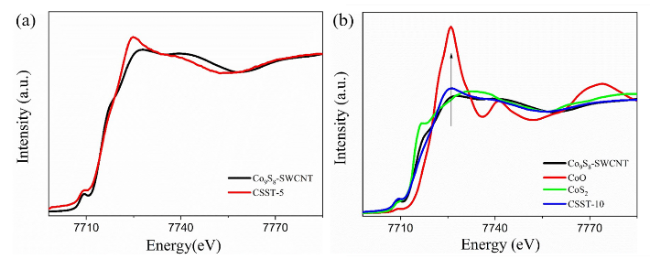
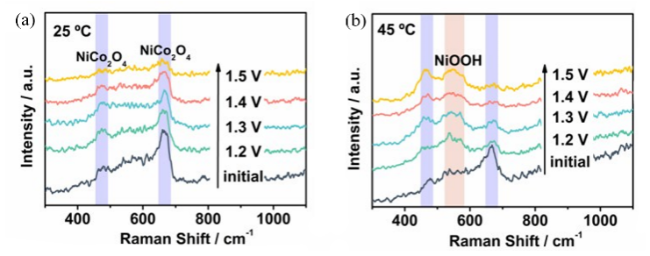
图3 归一化钴K边XANES谱。(a) Co9S8-单壁碳纳米管(Co9S8-SWCNT)及其在pH=5条件下OER反应后样品(CSST-5)的谱图;(b) Co9S8-SWCNT、CoO、CoS2及在pH=10条件下OER反应后样品(CSST-10)的谱图[50]Fig.3 Normalized Co K-edge XANES spectra. (a) Spectra of Co9S8-single walled carbon nanotube (Co9S8-SWCNT) and Co9S8-SWCNT after OER at pH=5 (CSST-5);(b) Spectra of Co9S8-SWCNT,CoO,CoS2 and Co9S8-SWCNT after OER at pH=10 (CSST-10)[50] |
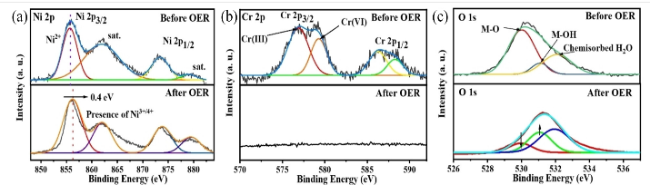
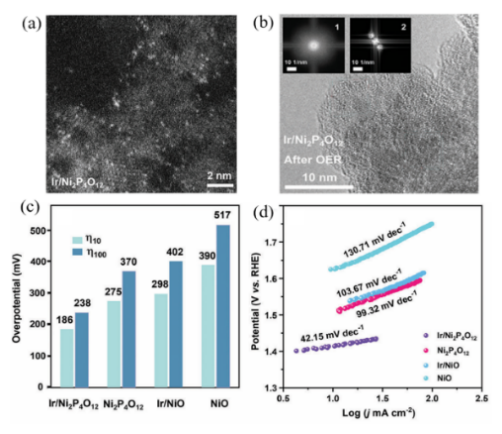
图9 (a) Ir/Ni2P4O12的HAADF-STEM图;(b) OER反应后Ir/Ni2P4O12的HRTEM图及选区FFT衍射图;(c) Ir/Ni2P4O12与其他催化剂的过电位对比图;(d) Ir/Ni2P4O12与其他催化剂的Tafel斜率图[65]Fig.9 (a) HAADF-STEM image of Ir/Ni2P4O12. (b) Post-OER HRTEM image and selected-area FFT pattern of Ir/Ni2P4O12. (c) Comparative overpotentials of Ir/Ni2P4O12 and other catalysts. (d) Tafel slope comparison for Ir/Ni2P4O12 and reference catalysts[65] |
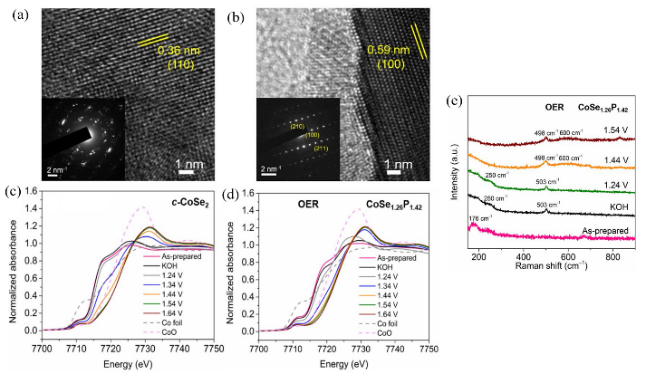
图12 (a) c-CoSe2的HRTEM及SAED图片;(b) CoSe1.26P1.42的HRTEM及SAED图片;(c,d) c-CoSe2 CoSe1.26P1.42的XANES图;(e) CoSe1.26P1.42的原位拉曼光谱图[74]Fig.12 (a) HRTEM image and SAED pattern of c-CoSe2;(b) HRTEM image and SAED pattern of CoSe1.26P1.42;(c,d) XANES spectra of c-CoSe2 and CoSe1.26P1.42;(e) in-situ Raman spectra of CoSe1.26P1.42[74] |
表1 不同催化剂重构前后OER性能对比Table 1 Comparison of OER performance before and after reconstruction with different catalysts |
| Reconstruction strategy | Pre-reconstruction | Post-reconstruction | OER performance improvement | Ref |
|---|---|---|---|---|
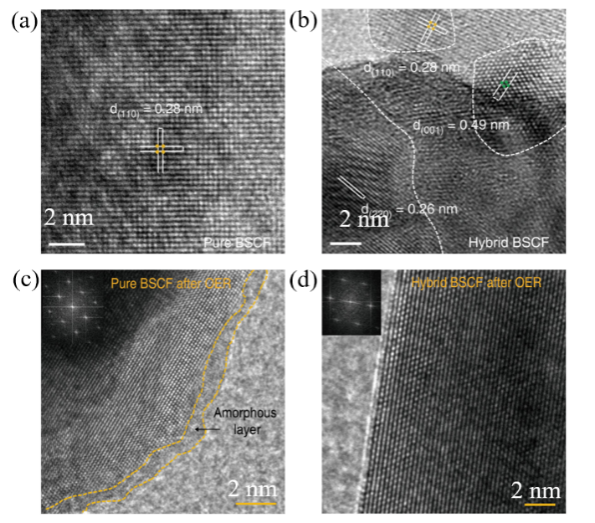
| Cation leaching | Ba0.35Sr0.65Co0.8Fe0.2O3-δ | Ba0.35Sr0.65Co0.8Fe0.2O3-δ;BaCl2;SrCl2 | η@10 mA/cm2:399→260 mV | 60 |
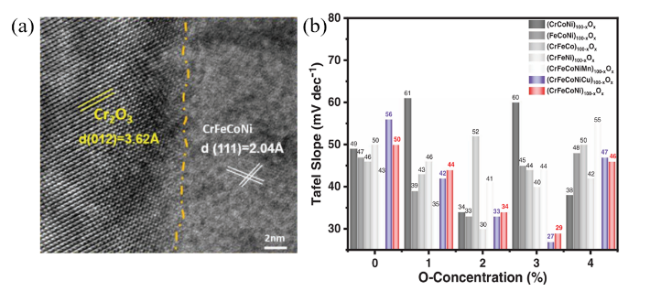
| CrFeCoNi high-entropy alloy | Cr2O3 microregions in alloy matrix | η@10 mA/cm2:258→196 mV | 61 | |
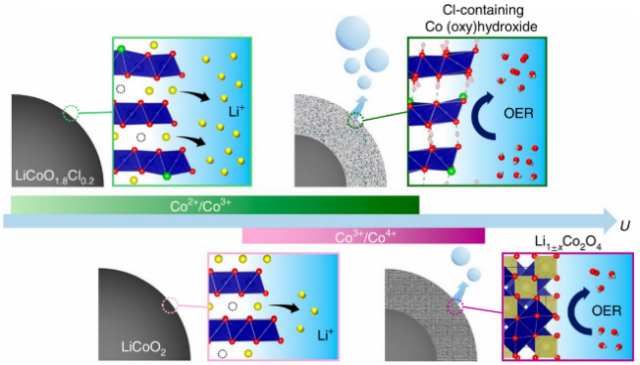
| LiCoO2-xClx | Amorphous Cl-doped Co (oxy)hydroxide | η@80 mA/cm2:290 mV | 62 | |
| Anion leaching | NiSe2/NiS2 | NiOOH with surface-adsorbed sulfate | j@500 mV:47→221 mA/cm2 | 64 |
| Ir/Ni2P4O12 | NiOOH | η@10 mA/cm2:275→186 mV | 65 | |
| SO42--loaded NiFe hydroxide | High-valence NiFeOOH | η@50 mA/cm2:296→234 mV | 66 | |
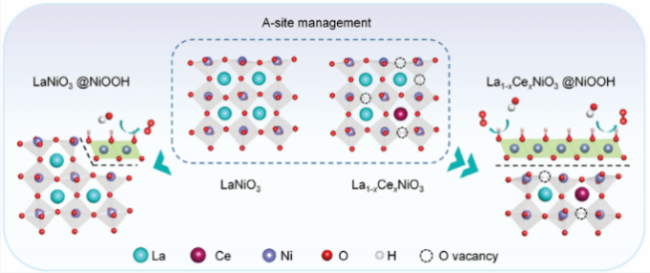
| Metal dopin | LaNiO3 | NiOOH | η@10 mA/cm2:460→270 mV | 69 |
| Pt/NiO | NiOOH | η@10 mA/cm2:510→358 mV | 70 | |
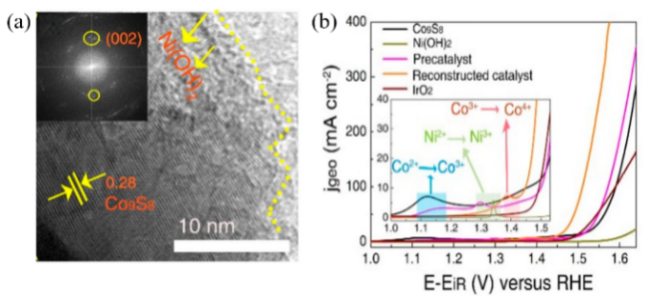
| CoS2@Ni(OH)2 | Fe-doped NiOOH/CoOOH heterostructure | η@50 mA/cm2:310→260 mV | 71 | |
| Non-metal doping | CoSe1.26P1.42 | CoOOH | η@10 mA/cm2:300→255 mV | 74 |
| SrCoO3-δ | SrCo0.95P0.05O3-δ | η@10 mA/cm2:520→461 mV | 75 | |
| Co-doping | Fe18.8W3.9B8.2P3.8O65.3 | W-FeOOH@FeB core-shell structure | η@10 mA/cm2:284→256 mV | 78 |
| Low-valence Ni in NiCoFeP oxyhydroxide | Ni4+-containing NiCoFeP oxyhydroxide | η@50 mA/cm2:588→330 mV | 79 | |
| Size modulation | NiO/Ni microparticles | Polycrystalline NiOOH nanosheets | η@10 mA/cm2:439→281 mV | 82 |
| N-doped C-supported NiMoFeO core-shell | NiFeOOH/NiFe | η@100 mA/cm2:400→290 mV | 83 |
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
(高秋璐, 刁鹏. 工程科学学报, 2025, 47(4): 923.)
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|
| [24] |
|
| [25] |
|
| [26] |
|
| [27] |
|
| [28] |
|
| [29] |
|
| [30] |
|
| [31] |
|
| [32] |
|
| [33] |
|
| [34] |
|
| [35] |
|
| [36] |
|
| [37] |
|
| [38] |
|
| [39] |
(孙正印, 向俊英, 叶壮, 章海霞. 功能材料, 2024, 55(12): 12192.)
|
| [40] |
(卢明龙, 张晓云, 杨帆, 王练, 王育乔. 化学进展, 2022, 34(3): 547.)
|
| [41] |
|
| [42] |
|
| [43] |
|
| [44] |
|
| [45] |
|
| [46] |
|
| [47] |
(廖凡, 陈子亮, 康振辉. 稀有金属, 2023, 47(1): 1.)
|
| [48] |
(薛世翔, 吴攀, 赵亮, 南艳丽, 雷琬莹. 化学进展, 2022, 34(12): 2686.)
|
| [49] |
|
| [50] |
|
| [51] |
|
| [52] |
|
| [53] |
|
| [54] |
|
| [55] |
|
| [56] |
|
| [57] |
|
| [58] |
|
| [59] |
(许文涛, 莫栩妍, 周洋, 翁祖贤, 莫坤玲, 吴炎桦, 蒋欣霖, 李丹, 蓝汤淇, 文欢, 郑伏琴, 樊友军, 陈卫. 物理化学学报, 2024, 40(8): 46.)
|
| [60] |
|
| [61] |
|
| [62] |
|
| [63] |
|
| [64] |
|
| [65] |
|
| [66] |
|
| [67] |
|
| [68] |
(黄琳尧, 罗密, 杨天华, 王晨光. 燃料化学学报(中英文), 2025, 53(3): 1.)
|
| [69] |
|
| [70] |
|
| [71] |
|
| [72] |
(雷航, 麦文杰. 科学通报, 2023, 68(4): 293.)
|
| [73] |
(张晓君, 马梁, 孙迎辉. 材料导报, 2021, 35(23): 23040.)
|
| [74] |
|
| [75] |
|
| [76] |
|
| [77] |
(杜茹雪, 滕慧雅. 化工科技, 2025, 33(2): 67.)
|
| [78] |
|
| [79] |
|
| [80] |
|
| [81] |
|
| [82] |
|
| [83] |
|
/
| 〈 |
|
〉 |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}