
The Single-Cell RNA Sequencing Technology Based on Microfluidic Chips
Received date: 2025-07-04
Revised date: 2025-08-05
Online published: 2025-08-29
Supported by
National Natural Science Foundation of China(32460624)
Kunming University of Science and Technology “Double First-Class” Science and Technology Project(202201BE070001-028)
Yunnan Provincial Education Science Planning Project(BC23002)
Technology 19th “Challenge Cup” National College Student Extracurricular Academic and Technological Works Competition Incubation Project(2024ZK128)
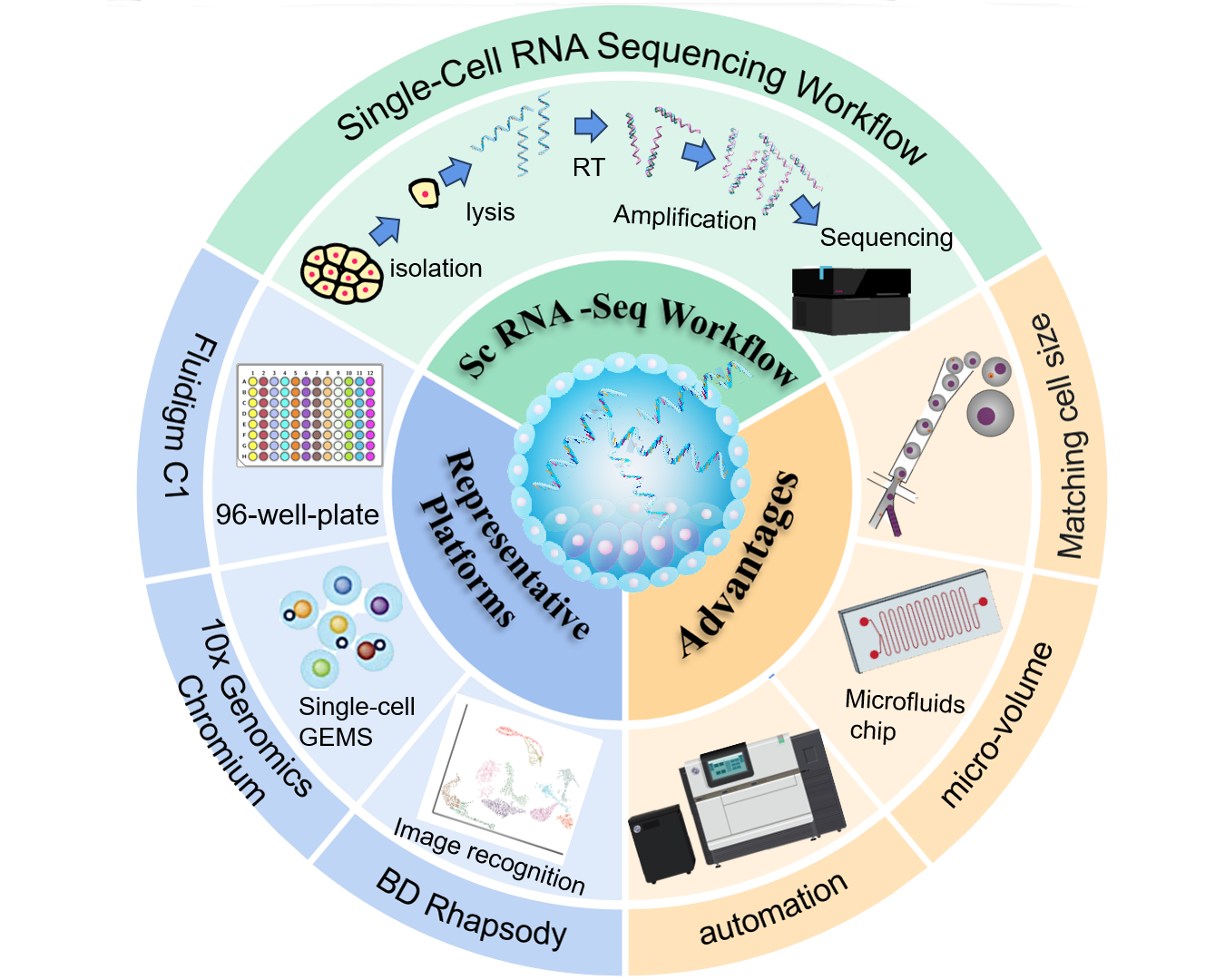
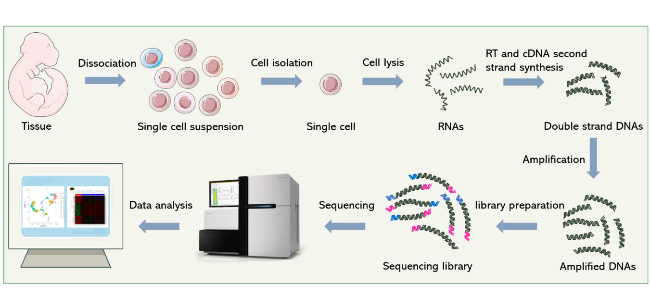
Cell heterogeneity is key to understanding life processes such as embryonic development and disease evolution,while traditional bulk cell RNA sequencing cannot resolve gene expression differences at the single-cell level. Although single-cell RNA sequencing (scRNA-seq) technology can construct transcriptomic maps at single-cell resolution,it faces challenges such as low efficiency in single-cell isolation and capture,and large deviations in trace RNA manipulation. Microfluidic chip technology,through a microscale fluid manipulation system,integrates processes such as single-cell isolation,lysis,reverse transcription,amplification,and sequencing library construction,achieving high-throughput,low sample loss,and automated operations,which significantly improve the efficiency and data reliability of scRNA-seq. This paper outlines the sequencing process of scRNA-seq,including steps such as single-cell isolation and capture,RNA extraction,reverse transcription and amplification,and single-cell sequencing. It analyzes the core advantages of microfluidic chips in adapting to single cells,precisely controlling reaction volumes,and realizing process automation,and briefly describes the technical principles and characteristics of representative platforms such as Fluidigm C1,10X Genomics Chromium,and BD Rhapsody. Microfluidic chip technology provides an efficient and precise technical platform for scRNA-seq. In the future,with the continuous optimization of chip design and the improvement of multi-omics integrated analysis capabilities,we expect it to play a more profound role in resolving complex biological systems,revealing disease mechanisms,and even promoting precision medicine.
Contents
1 Introduction
2 Single-cell RNA sequencing workflow
2.1 Isolation and capture of single cells
2.2 RNA extraction,reverse transcription and amplification
2.3 Single-cell sequencing
3 Single-cell RNA sequencing technology based on microfluidic chips
3.1 Development history of scRNA-seq based on microfluidic chips
3.2 Core advantages of microfluidic chips in scRNA-seq
4 Representative microfluidic single-cell RNA sequencing platforms
4.1 Fluidigm C1 platform
4.2 10X Genomics chromium platform
4.3 BD rhapsody platform
5 Summary and prospects
Luxi Shu , Yan Zhang . The Single-Cell RNA Sequencing Technology Based on Microfluidic Chips[J]. Progress in Chemistry, 2026 , 38(2) : 283 -297 . DOI: 10.7536/PC20250706
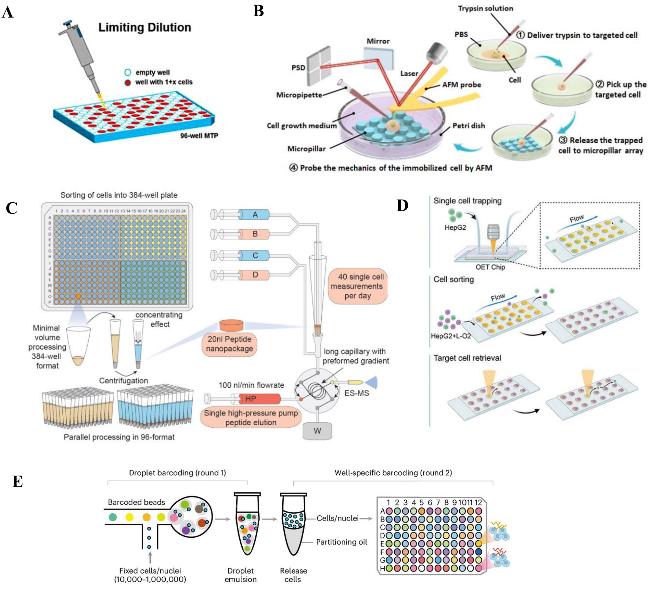
表1 常见的单细胞分离方法对比Table 1 Comparison of common single-cell isolation methods |
| Method | Principle | Throughput | Advantages | Limitations | Ref |
|---|---|---|---|---|---|
| Limited dilution | Based on Poisson distribution,single cells are randomly distributed in the well plate through gradient dilution (Fig.2A) | Low (≤ 384 well plates) | Simple equipment,low cost,suitable for small-scale research | High empty well rate,low throughput,dependent on operational experience | 20 |
| Micromanipulation | Manually manipulating a micropipette under a microscope to directly aspirate single cells (Fig.2B) | Extremely low | Precisely select cells of specific morphology or position without labeling interference | Extremely low throughput,time-consuming operation,dependent on highly skilled personnel | 21 |
| Fluorescence-activated cell sorting (FACS) | After fluorescent labeling,target single cells are sorted by droplet electric field (Fig.2C) | High (> 104 cells/hour) | High throughput,multi-parameter sorting (supporting combined labeling of multiple markers) | Expensive equipment,fluorescent labeling may change cell status,requiring high-quality single-cell suspensions | 22 |
| Micro-well technology | The size matches single cells,and cells randomly fall into the micro-wells (Fig.2D) | High (about 200 000 micro-wells) | High throughput,low damage,compatible with various cell types | The empty well rate is about 30 %,and the size of the micro-wells needs to strictly match the cell type | 23 |
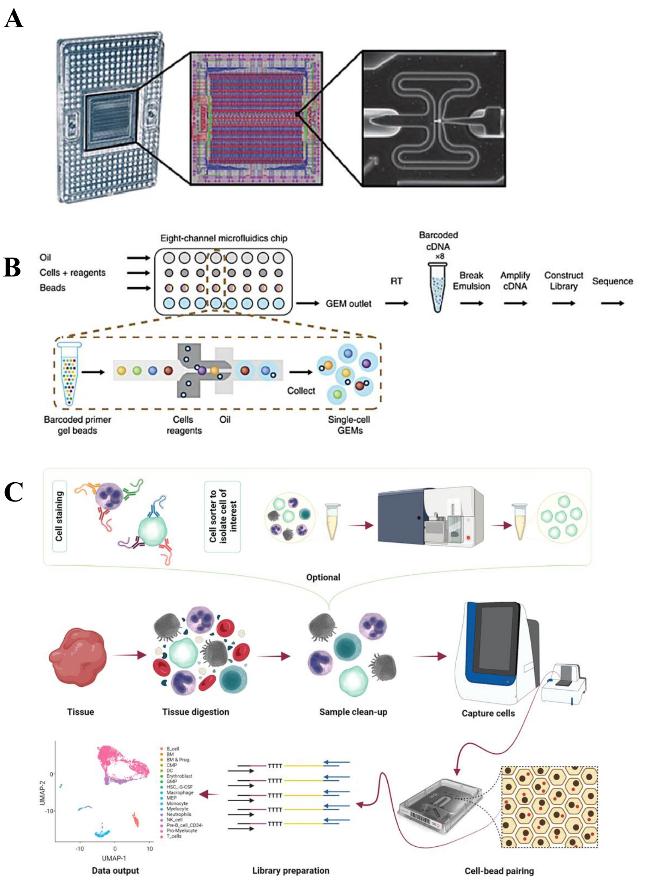
| Droplet microfluidics | Microfluidics generates independent droplets containing single cells + barcoded microbeads (Fig.2E) | Ultra-high (> 104 cells/time) | Ultra-high throughput,high capture efficiency,high degree of automation | High equipment and consumable costs,strict requirements for droplet stability,requiring complex bioinformatics for deduplication | 24 |
图2 常见的单细胞分离方法:(A) 有限稀释示意图[20];(B) 显微操作法分离单细胞[21];(C) FACS流程示意图[22];(D) 微孔列阵捕获单细胞[23];(E) 液滴微流控工作流程图[24]Fig.2 The most-used single-cell isolation methods. (A) Isolation of single cells by micromanipulation method[20];(B) schematic diagram of micromanipulation[21];(C) FACS flow diagram[22];(D) microporous arrays capture single cells[23];(E) diagram of droplet microfluidics workflow[24] |
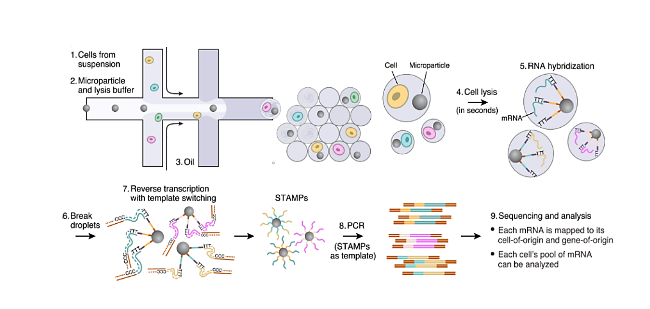
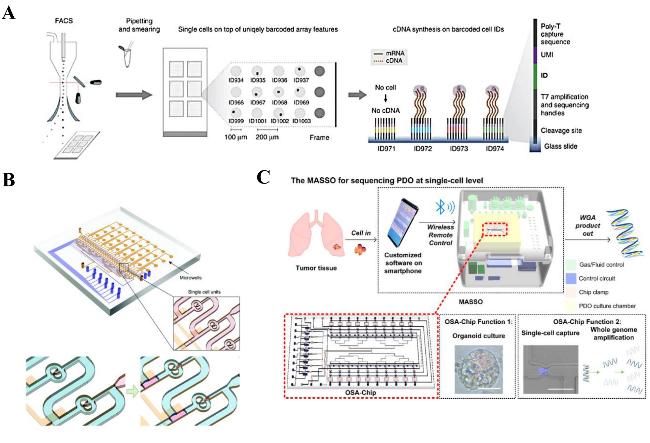
图4 (A) FACS机器将单细胞分选到带条形码的微阵列上,当单细胞落在条形码oligo-dTVN引物(ID)顶部时,才会转录cDNA[69];(B) 通过气动控制进行单细胞分离和皮升液滴生成的微流控芯片[75];(C) 基于微流控芯片的单细胞类器官自动测序系统示意图[84]Fig.4 (A) The FACS machine sorted single cells onto a barcoded microarray,and the cDNA was transcribed only when the single cells landed on top of the barcoded ogo-dTVN primer (ID)[69]. (B) Microfluidic chip for single cell isolation and picolitre droplet generation by pneumatic control[75]. (C) Schematic diagram of a Microfluidic chip-based Automatic System for Sequencing Organoids at the single-cell level[84] |
表2 代表性的微流控单细胞RNA测序平台Table 2 Representative microfluidic single-cell RNA sequencing platforms |
| Platform | Core Technology | Throughput | Core Advantages | Limitations | Applicable Samples | Ref |
|---|---|---|---|---|---|---|
| Fluidigm C1 | Microwell Array | Low | Stable automated workflow | Low throughput,high cost | Immune cells,neurons cell lines | 88 |
| 10X Genomics | Droplet Microfluidics | High | Standardized analysis,compatible with multiple species | Omission of expressed genes | Blood cells,tumor cells,organoids | 59 |
| BD Rhapsody | Magnetic Bead Encoding | High | Multi-omics integration | Require cell viability > 90% | Fresh immune cells,stem cells,early-stage embryos | 89 |
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|
| [24] |
|
| [25] |
|
| [26] |
|
| [27] |
|
| [28] |
|
| [29] |
|
| [30] |
|
| [31] |
|
| [32] |
|
| [33] |
|
| [34] |
|
| [35] |
|
| [36] |
|
| [37] |
|
| [38] |
|
| [39] |
|
| [40] |
|
| [41] |
|
| [42] |
|
| [43] |
|
| [44] |
|
| [45] |
|
| [46] |
|
| [47] |
|
| [48] |
|
| [49] |
|
| [50] |
|
| [51] |
|
| [52] |
|
| [53] |
|
| [54] |
|
| [55] |
|
| [56] |
|
| [57] |
|
| [58] |
|
| [59] |
|
| [60] |
|
| [61] |
|
| [62] |
|
| [63] |
|
| [64] |
|
| [65] |
|
| [66] |
|
| [67] |
|
| [68] |
|
| [69] |
|
| [70] |
|
| [71] |
|
| [72] |
|
| [73] |
|
| [74] |
|
| [75] |
|
| [76] |
|
| [77] |
|
| [78] |
|
| [79] |
|
| [80] |
|
| [81] |
|
| [82] |
|
| [83] |
|
| [84] |
|
| [85] |
|
| [86] |
|
| [87] |
|
| [88] |
|
| [89] |
|
| [90] |
|
| [91] |
|
| [92] |
|
| [93] |
|
| [94] |
|
| [95] |
|
| [96] |
|
| [97] |
|
| [98] |
|
| [99] |
|
| [100] |
|
| [101] |
|
| [102] |
|
| [103] |
|
| [104] |
|
| [105] |
|
| [106] |
|
| [107] |
|
| [108] |
|
/
| 〈 |
|
〉 |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}