
Peptides Secondary Structure of α-Sheet
Received date: 2025-06-20
Revised date: 2025-08-14
Online published: 2026-02-04
Supported by
National Natural Science Foundation of China(22372198)
National Natural Science Foundation of China(22402227)
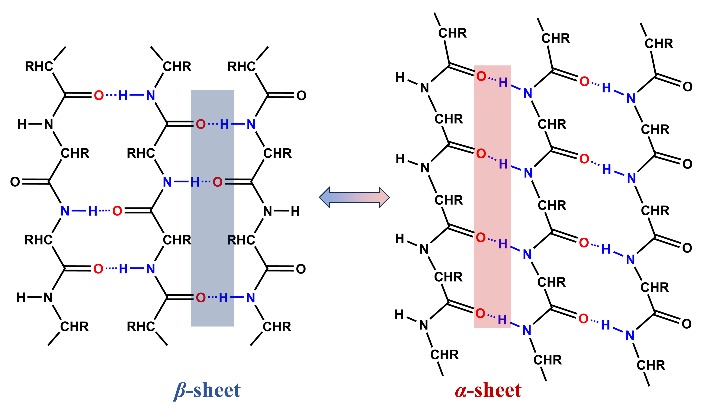
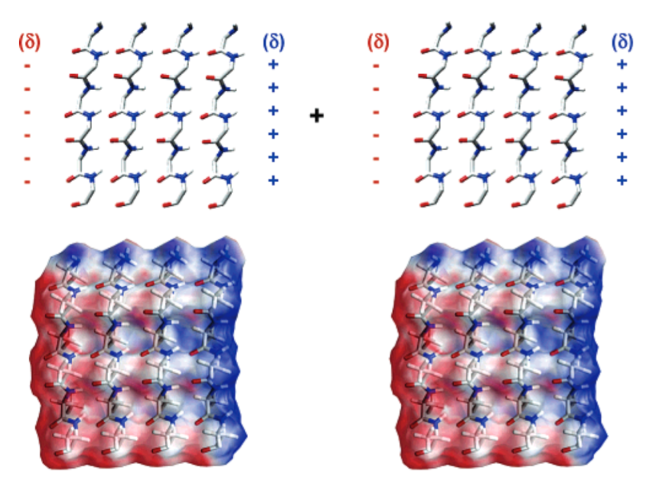
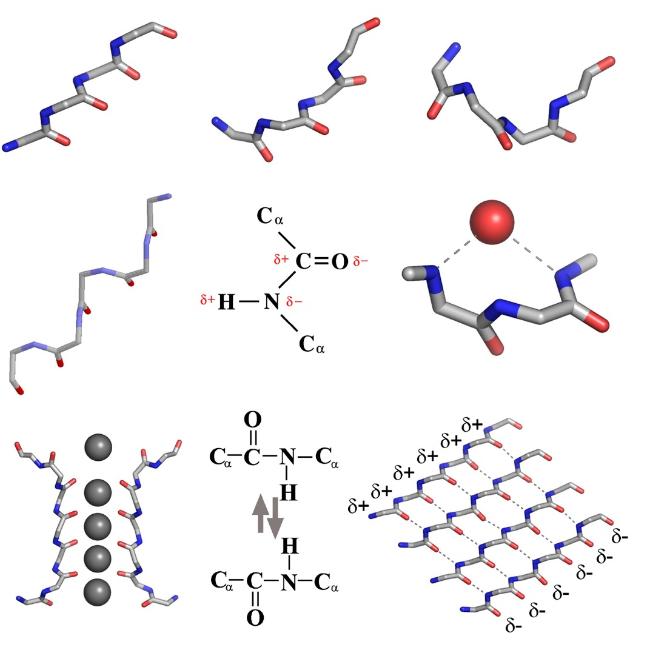
α-sheet is a rare secondary structure of peptides. Unlike common peptides secondary structures,α-sheet exhibits polarity with orderly arranged inter-strand hydrogen bonds while maintaining an extended conformation of α-strand. Due to its unstable molecular arrangement,it has long been ignored as a temporary product during the protein folding process. With the advancement of crystallography and molecular dynamics simulation technologies,research on amyloid proteins causing various neurodegenerative diseases has found that α-sheet might be a critical intermediate in the formation of amyloid fibrils. Therefore,defining the formation cause and assembly mechanism of α-sheet can help to further understand the pathogenic principle of amyloid-related diseases and propose early diagnosis and targeted treatment strategies,as well as help to design self-assembly peptide biomaterials with various functions,such as piezoelectricity,biomimetic catalysis and drug delivery. In this review,we summarize recent progress of the peptides secondary structure,especially the rare secondary structures led by α-sheet,and focus on reviewing the self-assembly mechanism,regulatory mode and supramolecular structure of α-sheet peptides. In addition,the development potential of biomaterials based on self-assembly peptides has also been discussed.
Contents
1 Introduction
2 Peptide secondary structure in neurodegenerative diseases
2.1 β-sheet amyloid fibril
2.2 α-sheet intermediate
2.3 α to β conformational change
2.4 α-sheet peptide targeted therapy
3 Self-assembly peptides based on different chirality
4 Self-assembly peptides based on different secondary structure
4.1 β-sheet
4.2 α-helix
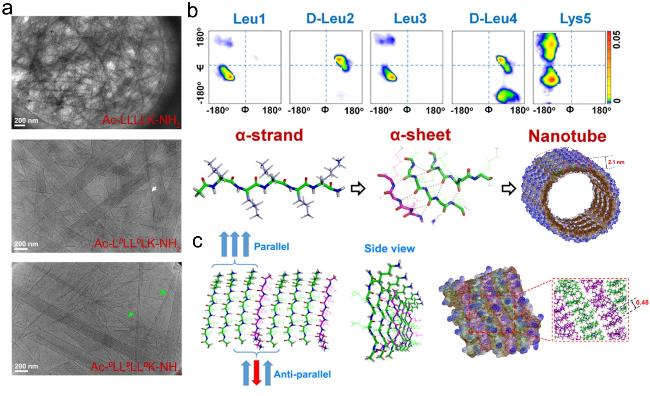
4.3 α-sheet
5 Conclusion and outlook
Zhaoyu Chen , Xiaoyue Ma , Henghao Yu , Hai Xu . Peptides Secondary Structure of α-Sheet[J]. Progress in Chemistry, 2026 , 38(2) : 319 -336 . DOI: 10.7536/PC20250612
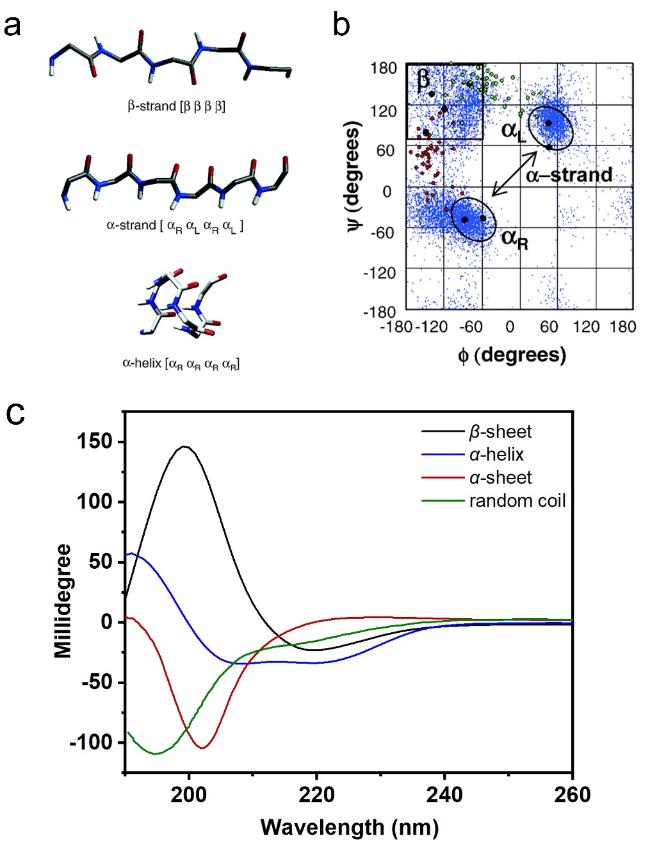
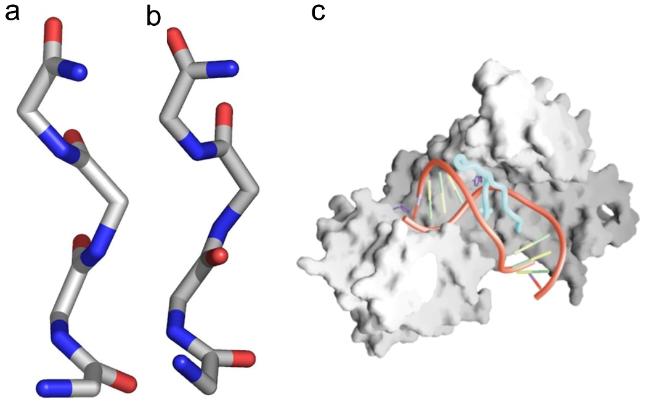
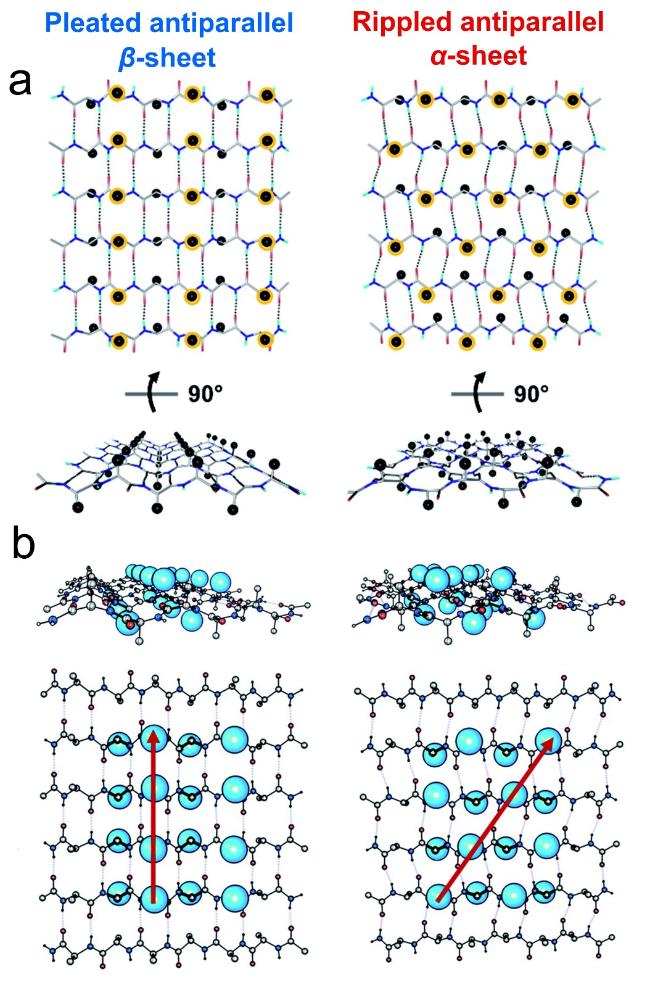
图2 α-折叠结构。(a)β-折叠链、α-折叠链和α-螺旋链的主链结构;(b)不同构象结构在拉氏图上的位置[34];(c)β-折叠、α-折叠、α-螺旋和无规卷曲的圆二色光谱图[13]Fig.2 α-sheet structure. (a) β-strand,α-strand,and α-helix’s main chain structures;(b) positions of the underlying local conformations of these structures on Ramachandran map[34];(c) CD spectra of β-sheet,α-sheet,α-helix and random coil[13]. Copyright 2006,American Chemical Society;Copyright 2022,American Chemical Society |
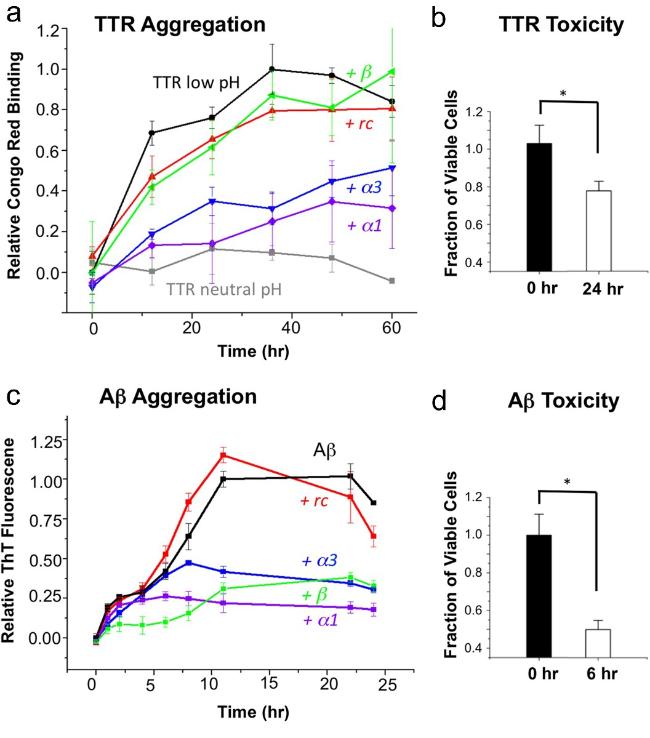
图3 α-折叠多肽抑制淀粉样蛋白形成并选择性结合毒性寡聚体示意图[35]:(a) 不同多肽与TTR蛋白共孵育后的聚集情况;(b) TTR溶液对神经肿瘤细胞SH-SY5Y的毒性;(c) 不同多肽与Aβ蛋白共孵育后的聚集情况;(d) Aβ溶液对SH-SY5Y的细胞毒性Fig.3 α-sheet peptides inhibit amyloid protein formation and selectively bind toxic species[35]. (a) Aggregation of different peptides after co-incubation with TTR. (b) The toxicity of TTR solution to neuroblastoma cell SH-SY5Y. (c) Aggregation of different peptides after co-incubation with Aβ. (d) The toxicity of Aβ to SH-SY5Y cell. Reproduced from Hopping et al. under the CC0 public domain dedication,eLife |
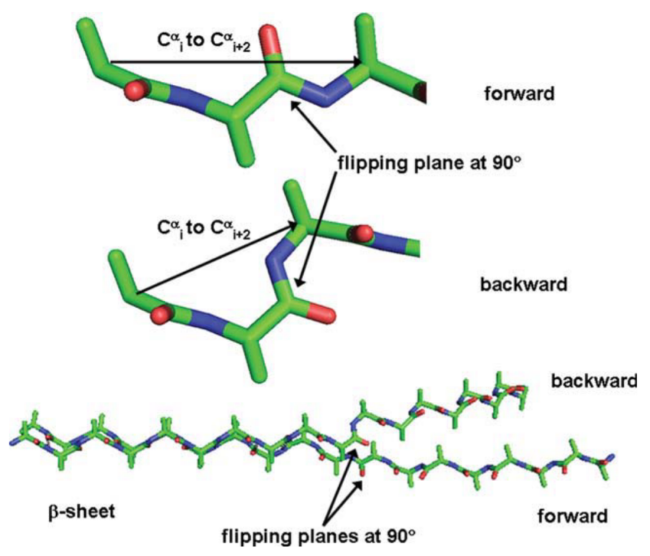
图5 UvrB-DNA解旋酶中的αRαL和ββ肽平面翻转[42-43]:(a) 主链呈αLαRαLαR构象,(b) 肽平面翻转180°局部呈现ββ构象,(c) UvrB-DNA解旋酶示意图Fig.5 αRαL to ββ peptide-plane flipping in UvrB DNA helicase[42-43]. (a) αLαRαLαR conformation backbone,(b) ββ conformation after 180° peptide plane flipping,(c) schematic diagram of UvrB-DNA helicase. Copyright 2006,Elsevier,Ltd |
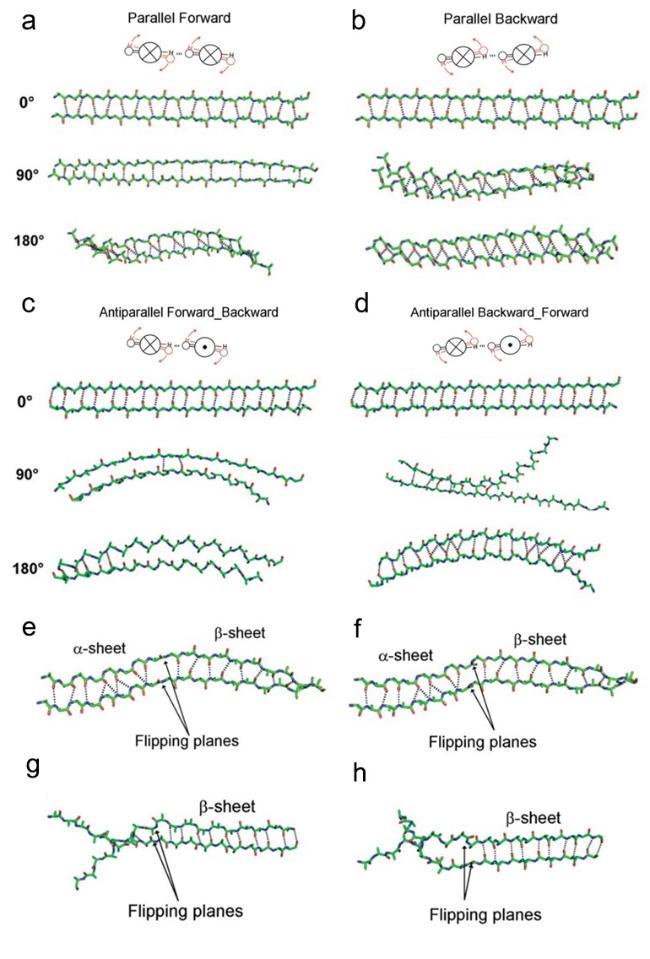
图7 α-折叠通过肽平面翻转向β-折叠构象转变模型[45]。协同翻转:(a) 平行向前,(b) 平行向后,(c) 反平行向前和(d) 反平行向后。顺序翻转:(e) 平行向前,(f) 平行向后,(g) 反平行向前和(h) 反平行向后Fig.7 α-sheet to β-sheet conformation transition through peptide plane flipping[45]. Concerted flipping:(a) parallel forward,(b) parallel backward;(c) antiparallel forward,(d) antiparallel backward. Sequential flipping:(e) parallel forward,(f) parallel backward;(g) antiparallel forward,(h) antiparallel backward. Copyright 2011,Wiley-Liss,Inc |
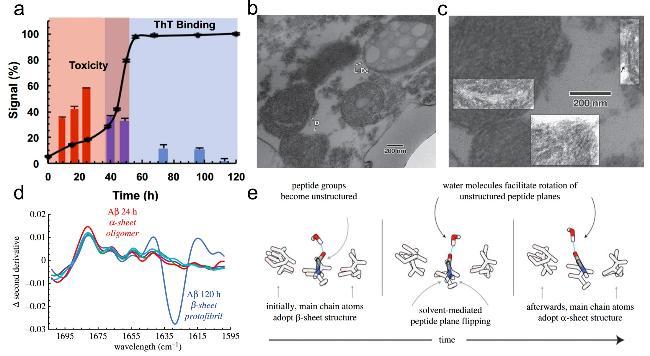
图8 α-折叠寡聚物的光谱与临床证据:(a) α-折叠细胞毒性实验[48],(b) 阿尔茨海默症大脑皮层细胞中浅色与深色包涵体,(c) 深色包涵体中的α-折叠寡聚物[50],(d) α-折叠MMS-IR特征光谱,(e) β-折叠向α-折叠转变示意图[49]Fig.8 Spectral and clinical evidence of α-sheet oligomers. (a) α-sheet cytotoxicity assay[48],(b) light and dark inclusions in Alzheimer-cerebral cortex,(c) α-sheet oligomers in dark inclusions[50],(d) α-sheet MMS-IR spectrum,(e) schematic diagram of the transition from β-sheet to α-sheet[49]. Copyright 2019,National Academy of Sciences;Copyright 2022,Sage Publications |
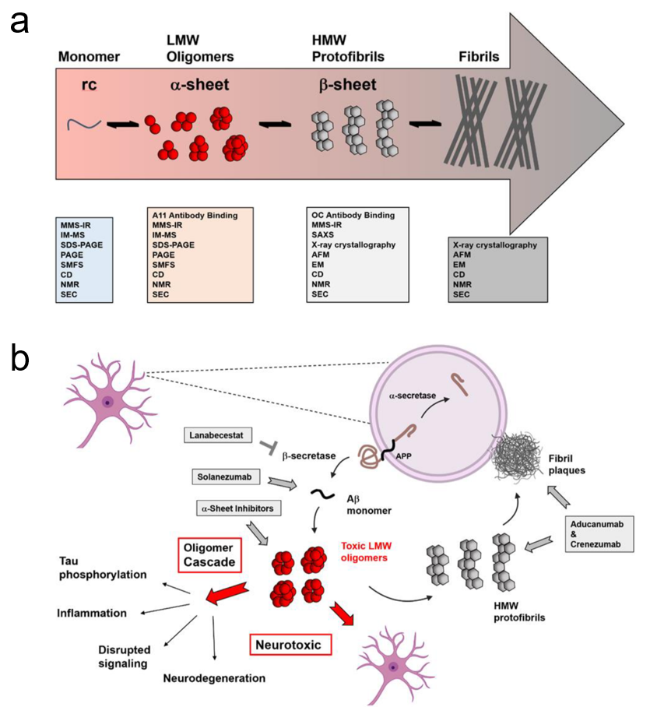
图9 神经退行性疾病过程中的构象转变及治疗策略[31]:(a)构象转变过程及表征技术,(b) 靶向药物及作用位点Fig.9 Conformational changes and treatment strategies in neurodegenerative diseases[31]. (a) Conformational transition processes and characterization techniques,(b) targeted drugs and their action sites. Adapted from Shea and Daggett under the CC BY license,MDPI |
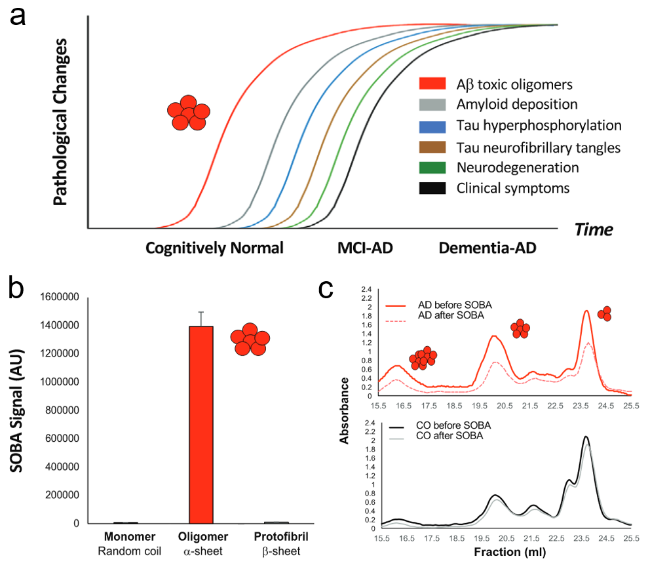
图10 阿尔茨海默症的分子病理学过程[32]:(a) 不同时间阶段阿尔茨海默症的病理学事件,(b) α-折叠寡聚体表现出明显的SOBA信号,(c) 经过SOBA治疗后的AD患者血液中α-折叠寡聚体信号明显下降Fig.10 The molecular pathology of Alzheimer's disease[32]. (a) Pathological changes in Alzheimer's disease at different stages,(b) α-sheet oligomers show obvious SOBA signals,(c) after SOBA treatment,the α-sheet oligomer signal in the blood of AD patients decreased significantly. Copyright 2022,National Academy of Sciences |
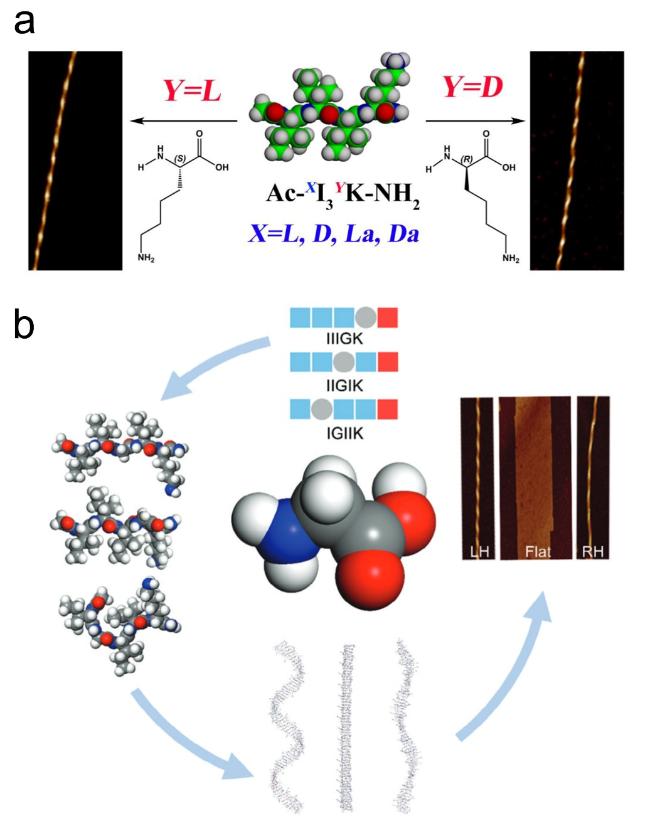
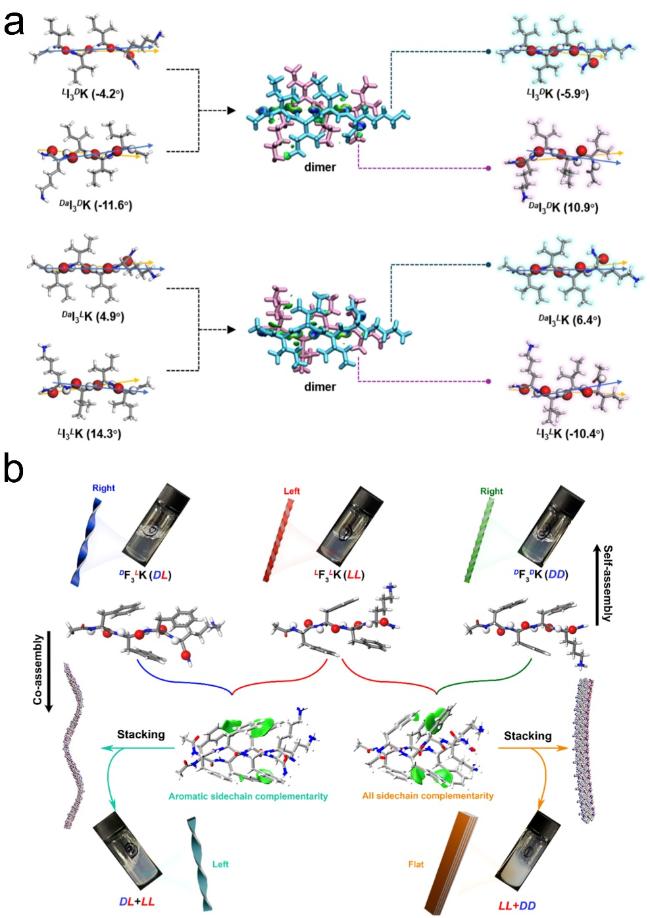
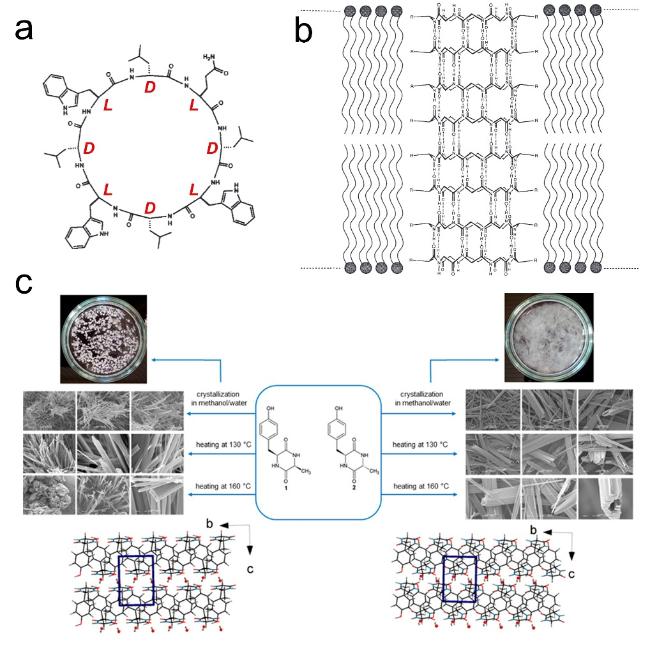
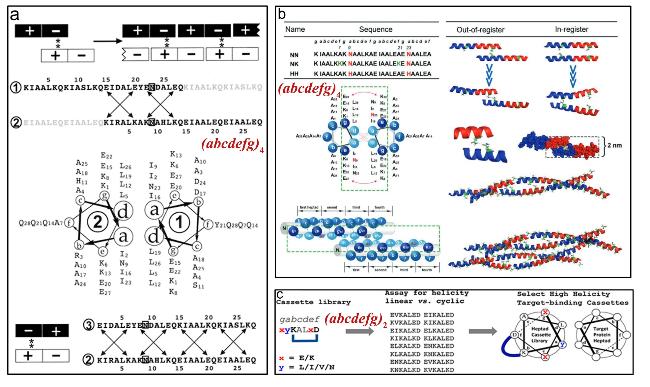
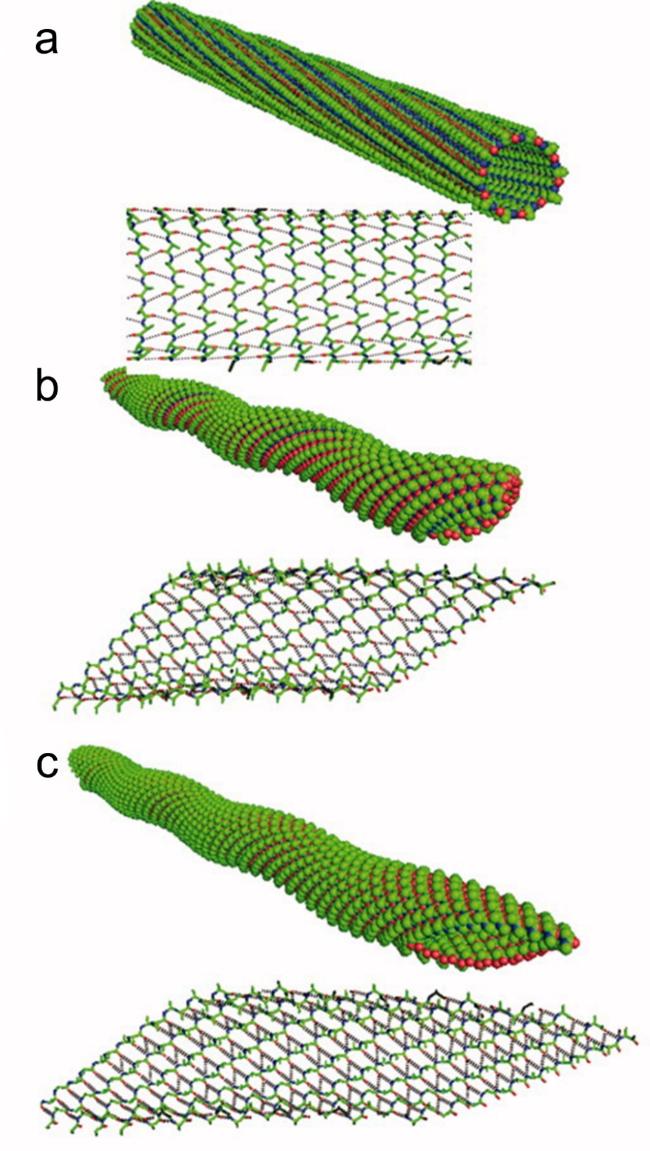
图14 环肽序列设计:(a,b) 环肽cyclo-[(LTrp-DLeu)3- LGln-DLeu]序列及其轴向组装形成的纳米孔道[69],(c) cyclo- (LTyr-LAla)和cyclo-(LTyr-DAla)各自组装形貌示意图[71]Fig.14 Cyclopeptide sequence design:(a,b) Cyclopeptide cyclo-[(LTrp-DLeu)3-LGln-DLeu] sequence and its axial assembly forming a nanopore channel[69],(c) schematic diagram of the assembly morphology of cyclo-(LTyr-LAla) and cyclo-(LTyr-DAla)[71]. Copyright 1994,Springer Nature Limited;Copyright 2015,American Chemical Society |
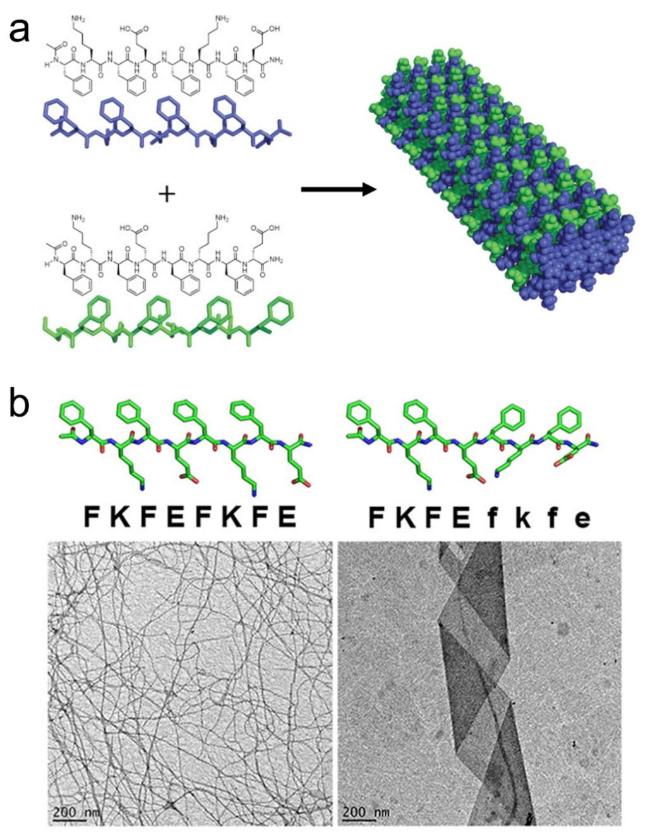
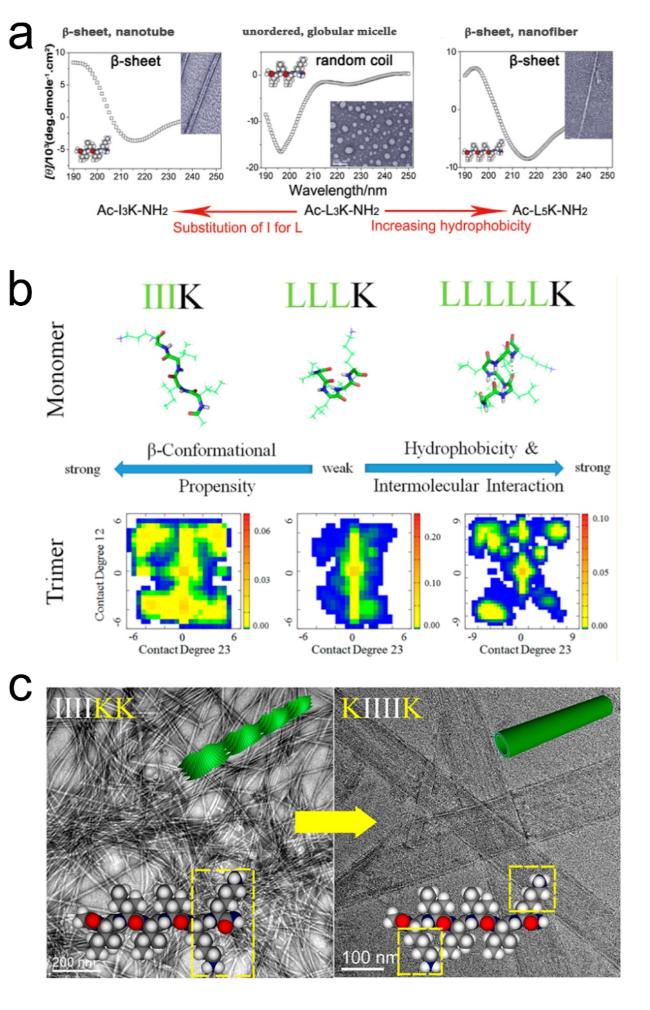
图15 β-折叠两亲性自组装超短肽的序列调控方式:(a,b) 氢键与疏水作用调控[82-83],(c) 氨基酸位置替换[84]Fig.15 Sequence regulation of β-sheet amphiphilic self-assembly short peptides:(a,b) regulation by hydrogen bonding and hydrophobic interactions[82-83],(c) amino acid substitution[84]. Copyright 2011,Wiley;Copyright 2016,American Chemical Society |
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|
| [24] |
|
| [25] |
|
| [26] |
|
| [27] |
|
| [28] |
|
| [29] |
|
| [30] |
|
| [31] |
|
| [32] |
|
| [33] |
|
| [34] |
|
| [35] |
|
| [36] |
|
| [37] |
|
| [38] |
|
| [39] |
|
| [40] |
|
| [41] |
|
| [42] |
|
| [43] |
|
| [44] |
|
| [45] |
|
| [46] |
|
| [47] |
|
| [48] |
|
| [49] |
|
| [50] |
|
| [51] |
|
| [52] |
|
| [53] |
|
| [54] |
|
| [55] |
|
| [56] |
|
| [57] |
|
| [58] |
|
| [59] |
|
| [60] |
|
| [61] |
(林代武, 邢起国, 王跃飞, 齐崴, 苏荣欣, 何志敏. 化学进展, 2019, 31(12): 1623.)
|
| [62] |
(王继乾, 闫宏宇, 李洁, 张丽艳, 赵玉荣, 徐海. 化学进展, 2018, 30(8): 1121.)
|
| [63] |
|
| [64] |
|
| [65] |
|
| [66] |
|
| [67] |
|
| [68] |
|
| [69] |
|
| [70] |
|
| [71] |
|
| [72] |
|
| [73] |
|
| [74] |
|
| [75] |
|
| [76] |
|
| [77] |
|
| [78] |
|
| [79] |
|
| [80] |
|
| [81] |
|
| [82] |
|
| [83] |
|
| [84] |
|
| [85] |
|
| [86] |
|
| [87] |
|
| [88] |
|
| [89] |
(苗君萍, 张昭乾, 辛少鹏, 胡云霞. 化学进展, 2025, 37(2): 195.)
|
| [90] |
|
| [91] |
|
/
| 〈 |
|
〉 |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}