
Signal Amplification Mechanisms in Photoelectrochemical Biosensors: From Photoelectric Conversion to Signal Output
† These authors contributed equally to this work
Received date: 2026-01-04
Revised date: 2026-01-29
Online published: 2026-03-18
Supported by
National Natural Science Foundation of China(22474124)
National Natural Science Foundation of China(21475116)
National Natural Science Foundation of China(21575125)
Project for Yangzhou City and Yangzhou University corporation(YZ2023204)
Open Research Fund of State Key Laboratory of Analytical Chemistry for Life Science(SKLACLS2405)
Postgraduate Research & Practice Innovation Program of Jiangsu Province(KYCX25_3936)
Jiangsu Provincial Key Laboratory of Green & Functional Materials Environmental Chemistry
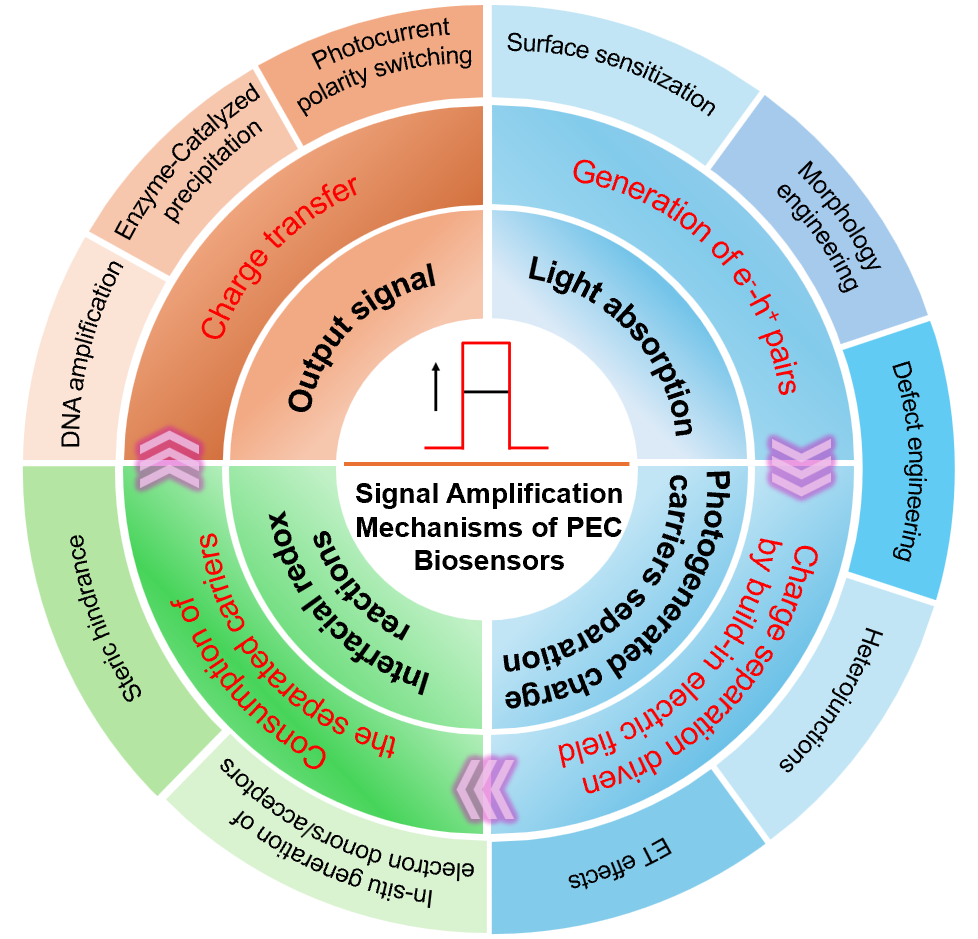
Photoelectrochemical (PEC) biosensors, as an emerging analytical platform, offer significant advantages, including low background signals, high sensitivity, and operational simplicity, due to the inherent separation of the excitation source and the detection signal. The core of achieving high performance in PEC biosensors lies in the development of efficient signal amplification strategies. This review systematically summarizes recent research progress on signal amplification mechanisms in PEC biosensors. Photoelectric †conversion constitutes the basis of PEC sensing, primarily involving three essential processes: light harvesting, charge carrier separation, and interfacial reaction. Based on this, the prevailing signal amplification mechanisms are reviewed from the core processes of photoelectric conversion to the design of signal output. Simultaneously, the design principles and characteristics of these mechanisms are delved. Finally, this review examines the challenges of PEC sensing technologies and explores future trends. This review aims to provide theoretical guidance for the rational design of high-performance PEC biosensors and to promote their further development in applications of analysis.
1 Introduction
2 Signal amplification mechanisms in PEC sensors
2.1 Modulating light absorption and photogenerated charge carriers separation
2.2 Modulating interfacial redox reactions
2.3 Modulating the output signal
3 Challenges and perspectives
Sitian Long , Haibing Zhu , Yuchen Du , Yadong Xue , Juan Li , Zhanjun Yang . Signal Amplification Mechanisms in Photoelectrochemical Biosensors: From Photoelectric Conversion to Signal Output[J]. Progress in Chemistry, 2026 , 38(3) : 532 -560 . DOI: 10.7536/PC20260101
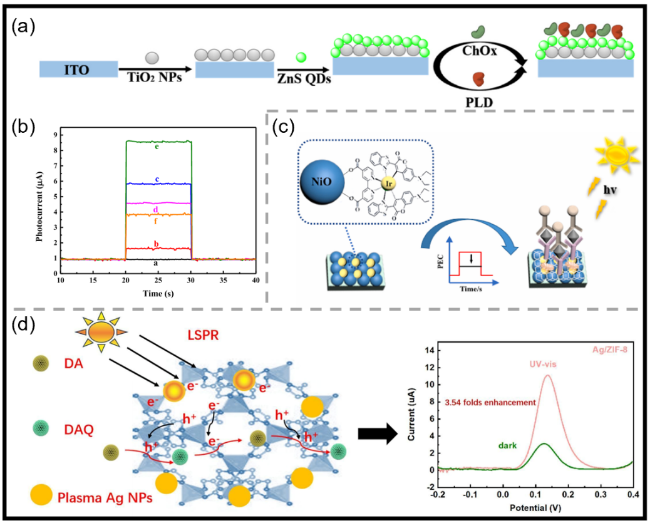
图2 (a)ITO/TiO2 NPs/ZnS QDs/胆碱氧化酶(ChOx)/磷脂酶D(PLD)电极的制备过程示意图[42];(b)裸ITO(a)、ITO/TiO2 NPs(b)、ITO/TiO2 NPs/ZnS QDs(c)、ITO/TiO2 NPs/ZnS QDs/ChOx(d)和ITO/TiO2 NPs/ZnS QDs/ChOx/PLD(e)的光电流响应曲线,PEC测试在含有1 mmol/L PC在PBS(10 mmol/L)中进行,偏置电压为0.4 V;ITO/TiO2 NPs/ZnS QDs/ChOx/PLD(f)在不含1 mmol/L PC在PBS(10 mmol/L)中、偏置电压0.4 V条件下的光电流响应曲线[42];(c)[(C6)2Ir(dcbpy)]+敏化NiO光电阴极用于前列腺特异性抗原(PSA)检测的组装过程示意图[52];(d)基于Ag/ZIF-8复合材料检测DA的PEC传感机制示意图,以及Ag/ZIF-8电极在有光和无光条件下的DPV曲线[58]Fig.2 (a) Fabrication process of the ITO/TiO2 NPs/ZnS QDs/choline oxidase (ChOx)/phospholipase D (PLD) electrode[42]. Copyright 2025, Elsevier. (b) Photocurrent responses of bare ITO (a), ITO/TiO2 NPs (b), ITO/TiO2 NPs/ZnS QDs (c), ITO/TiO2 NPs/ZnS QDs/ChOx (d), and ITO/TiO2 NPs/ZnS QDs/ChOx/PLD (e), PEC measurements were carried out in PBS (10 mmol/L) with 1 mmol/L PC under 0.4 V bias voltage. Photocurrent responses of ITO/TiO2 NPs/ ZnS QDs/ChOx/PLD (f), in absence of 1 mmol/L PC in PBS (10 mmol/L) under 0.4 V bias voltage[42]. Copyright 2025, Elsevier. (c) Schematic diagram of the assembly process about [(C6)2Ir(dcbpy)]+-sensitized NiO photocathode for prostate-specific antigen (PSA) detection[52]. Copyright 2022, Elsevier. (d) Schematic diagram of the PEC sensing mechanism for DA detection using the Ag/ZIF-8 composite. And DPV curves of the Ag/ZIF-8 electrode in the presence and absence of light[58]. Copyright 2025, Elsevier |
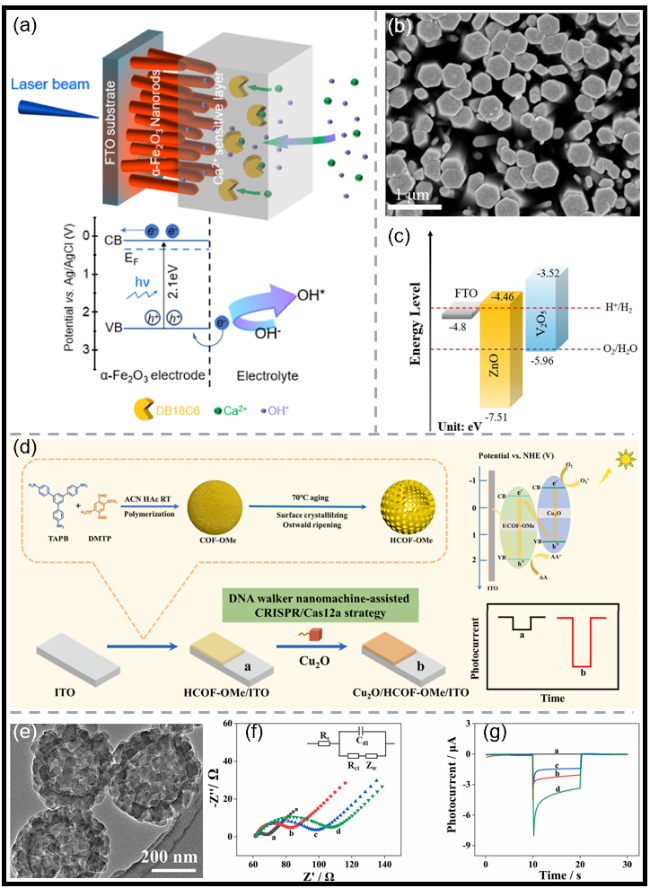
图3 (a)用于检测Ca2+ PEC传感的改性赤铁矿纳米棒示意图[65];(b)原始ZnO NRs的SEM俯视图像[68];(c)原始ZnO、ZnO/V2O5及V2O5的能级排列示意图[68];(d)基于空心结构HCOF-OMe构建的用于微囊藻毒素-LR(MC-LR)检测的PEC生物传感平台示意图[71];(e)实心COF-OMe的TEM图像[71];(f,g)不同修饰电极的Nyquist图及拟合等效电路图(f)与光电流响应曲线(g):(a)ITO、(b)HCOF-OMe/ITO、(c)GA/HCOF-OMe/ITO和(d)Cu2O/HCOF-OMe/ITO[71]Fig.3 (a) Schematic diagram of the modified hematite NRs for the PEC sensing of Ca2+[65]. Copyright 2022, American Chemical Society. (b) Top-view SEM image of the pristine ZnO NRs[68]. Copyright 2026, Elsevier. (c) Energy level alignment of the pristine ZnO, ZnO/V2O5, and V2O5[68]. Copyright 2026, Elsevier. (d) Schematic illustration of the proposed PEC biosensing platform for microcystin-LR (MC-LR) assay based on hollow structured HCOF-OMe[71]. Copyright 2025, Elsevier. (e) TEM image of the solid COF-OMe[71]. Copyright 2025, Elsevier. (f, g) Nyquist plots with fitted equivalent circuit diagram (f) and photocurrent responses (g) of different electrodes: (a) ITO, (b) HCOF-OMe/ITO, (c) GA/ HCOF-OMe/ITO, and (d) Cu2O/HCOF-OMe/ITO[71]. Copyright 2025, Elsevier |
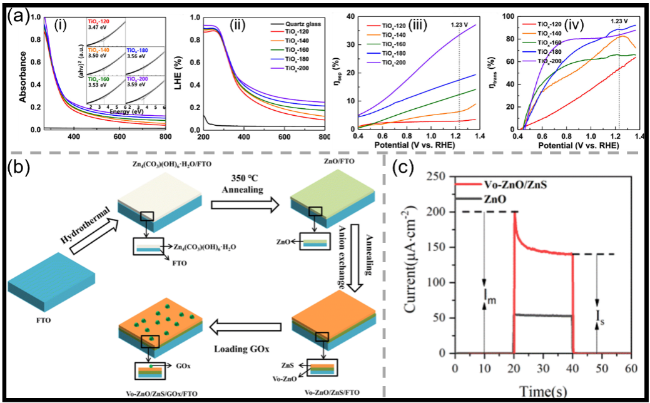
图4 (a)ALD-TiOx薄膜的(i)紫外-可见吸收光谱及(内插)Tauc图、(ii)光捕获效率曲线、(iii)电荷分离效率及(iv)电荷转移效率[72];(b)Vo-ZnO/ZnS/FTO电极的制备流程及葡萄糖传感器构建示意图[76];(c)ZnO与Vo-ZnO/ZnS的光电响应曲线[76]Fig.4 (a) (i) UV-vis absorbance spectra and (inset) Tauc plot, (ii) light-harvesting efficiency curves, (iii) charge separation efficiencies, and (iv) charge transfer efficiencies of ALD-TiOx thin films[72]. Copyright 2023, American Chemical Society. (b) Simple schematic of the preparation process of the Vo-ZnO/ZnS/FTO electrodes and glucose sensor preparation[76]. Copyright 2023, The Royal Society of Chemistry. (c) Photoelectric responses of the ZnO and Vo-ZnO/ZnS[76]. Copyright 2023, The Royal Society of Chemistry |
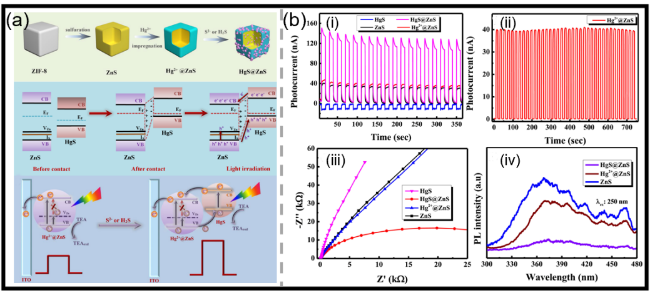
图5 (a)Hg2+@ZnS的制备过程、添加S2-或H2S后原位形成HgS@ZnS样品的示意图;复合前ZnS与HgS的能带结构排列,以及复合后在模拟太阳光照射下形成的具有Type-II异质结构的HgS@ZnS相应的电荷转移过程,以及基于Hg2+@ZnS的PEC传感器在模拟太阳光照射下对痕量S2-或H2S进行“信号增强”型分析的传感机理示意图[78];(b)(i)ZnS、Hg2+@ZnS、HgS@ZnS和HgS样品的光电流响应曲线,(ii)Hg2+@ZnS在750 s光开关循环测试下的光电流稳定性,(iii)所得样品的电化学阻抗谱,(iv)ZnS和HgS@ZnS样品的稳态光致发光(PL)光谱及时间分辨PL衰减曲线[78]Fig.5 (a) Schematic illustration of the preparation procedure of Hg2+@ZnS and in-situ formed HgS@ZnS samples after the addition of S2- or H2S, and the band structure alignments of ZnS and HgS before combination and the corresponding charge transferring of HgS@ZnS with Type-II heterostructure formed after the combination under simulated sunlight irradiation, and the sensing mechanism of Hg2+@ZnS-based PEC sensor for the “signal-on” analysis of trace S2- or H2S under simulated sunlight irradiation[78]. Copyright 2025, Elsevier. (b) (i) The photocurrent responses of ZnS, Hg2+@ZnS, HgS@ZnS and HgS samples, (ii) the photocurrent stability tests of Hg2+@ZnS under on/off light cycle test for 750 s. (iii) the electrochemical impedance spectra of so-obtained samples. (iv) the steady-state photoluminescence (PL) spectra and time-resolved PL decay of ZnS and HgS@ZnS samples[78]. Copyright 2025, Elsevier |
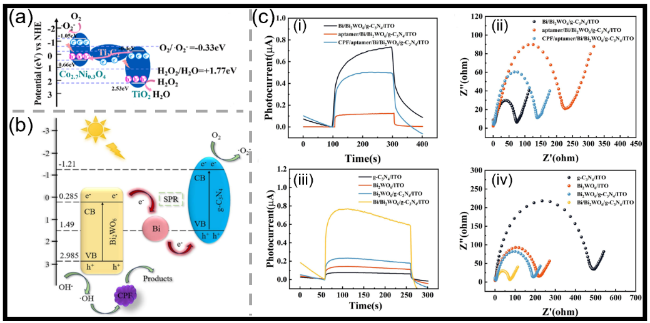
图6 (a)PEC传感器检测H2O2的作用机理示意图[81];(b)毒死蜱(CPF)检测中可能的Z型电子转移机理示意图[85];(c)(i)不同修饰电极的光电流响应曲线及(ii)Nyquist图:Bi/Bi2WO6/g-C3N4/ITO、aptamer/Bi/Bi2WO6/g-C3N4/ITO和CPF/aptamer/Bi/Bi2WO6/g-C3N4/ITO;(iii)g-C3N4/ITO、Bi2WO6/ITO、Bi2WO6/g-C3N4/ITO和Bi/Bi2WO6/g-C3N4/ITO的光电流响应对比及(iv)电化学阻抗谱(EIS)图[85]Fig.6 (a) Detection mechanism of H2O2 at the PEC sensor[81]. Copyright 2025, Elsevier. (b) Chlorpyrifos (CPF) detection of a possible Z-type electron transfer mechanism[85]. Copyright 2026, Elsevier. (c) (i) Photocurrent responses and (ii) Nyquist plots of different modified electrodes: Bi/Bi2WO6/g-C3N4/ITO, aptamer/Bi/Bi2WO6/g-C3N4/ITO, and CPF/aptamer/Bi/Bi2WO6/g-C3N4/ITO. (iii) Comparative photocurrent responses and (iv) electrochemical impedance spectroscopy (EIS) spectra of g-C3N4/ITO, Bi2WO6/ITO, Bi2WO6/g-C3N4/ITO, and Bi/Bi2WO6/g-C3N4/ITO[85]. Copyright 2026, Elsevier |
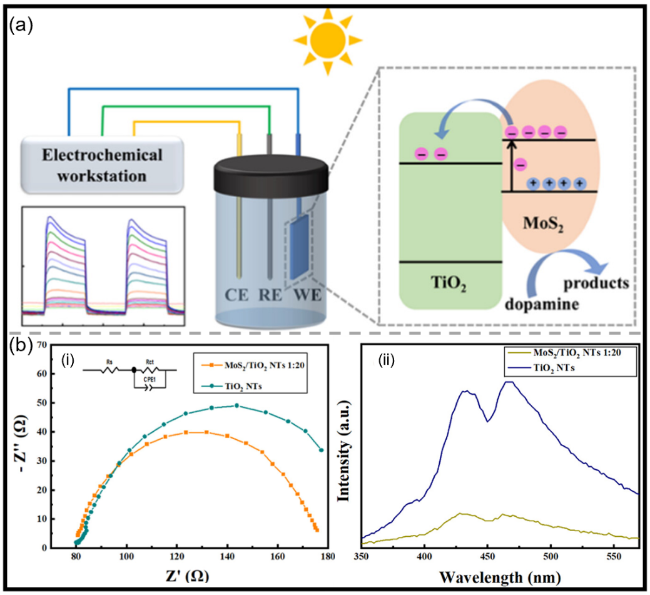
图7 (a)MoS2/TiO2 NTs p-n异质结可能的电子转移机理示意图;(b)MoS2/TiO2 NTs(1∶20)与TiO2 NTs的(i)EIS图谱及(ii)PL光谱[86]Fig.7 (a) A possible MoS2/TiO2 NTs p-n heterojunction electron transfer mechanism. (b) EIS plots (i), and PL spectrum (ii) of the MoS2/TiO2 NTs 1∶20 and TiO2 NTs[86]. Copyright 2025, American Chemical Society |
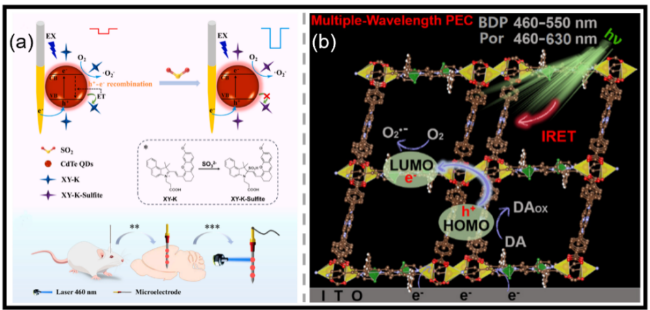
图8 (a)基于ET调制的PEC微传感器用于SO2检测及体内分析的示意图,采用光寻址微电极(*:XY-K的识别过程;**:体内靶标识别;***:体外信号检测)[94];(b)在存在DA和溶解氧的条件下,具有框架内IRET机制的dpMOF的PEC过程示意图[97]Fig.8 (a) The schematic illustration of the ET modulated PEC microsensor for the detection of SO2 and in vivo assays using the light addressable microelectrode (*: recognition process of XY-K; **: in vivo target recognizing; ***: in vitro signal detecting)[94]. Copyright 2024, Elsevier. (b) A schematic illustration of the PEC process of dpMOF with IRET mechanism in the presence of DA and dissolved O2[97]. Copyright 2025, American Chemical Society |
图9 (a)用于γH2AX检测的分裂型PEC免疫分析示意图[102];(b)夹心型免疫反应及集成了自供能PEC与比色免疫分析的化学氧化还原循环生物传感器构建示意图[103];(c)用于CIP检测的PEC适配体传感器构建及可能的电子转移机理示意图[104];(d)Ag2S QDs与Bi2S3的合成及Ag2S/Bi2S3/Au-ALP PEC传感电极的制备流程示意图[105];(e)(i)ITO/Ag2S/Au-ALP、(ii)ITO/Bi2S3/Au-ALP和(iii)ITO/Ag2S/Bi2S3/Au-ALP在(a)不含APP及(b)含0.05 mol/L AAP的0.01 mol/L Tris-HCl缓冲液(pH=8.2)中的光电流响应;(iv)ITO/Ag2S/Bi2S3/Au-ALP在(a)含0.05 mol/L AAP的0.01 mol/L Tris-HCl缓冲液(pH=8.2)及(b)在含0.05 mol/L AAP和50 nmol/L NaF的0.01 mol/L Tris-HCl缓冲液(pH=8.2)中的光电流响应[105]Fig.9 (a) Schematic illustration of the split-type PEC immunoassay for γH2AX detection[102]. Copyright 2023, Elsevier. (b) Schematic illustration on the fabrication of the sandwich-type immunoreaction and the designed chemical redox cycling biosensor, which integrated self-powered PEC and colorimetric immunoassay[103]. Copyright 2025, American Chemical Society. (c) Fabrication of PEC aptasensors for CIP detection, and the possible mechanism of electron transfer[104]. Copyright 2025, Elsevier. (d) Synthesis of Ag2S QDs and Bi2S3 and the fabrication of the Ag2S/Bi2S3/Au-ALP PEC sensing electrode[105]. Copyright 2024, American Chemical Society. (e) Photocurrent responses of (i) ITO/Ag2S/Au-ALP, (ii) ITO/Bi2S3/Au-ALP, and (iii) ITO/Ag2S/Bi2S3/Au-ALP in (a) 0.01 mol/L Tris-HCl (pH 8.2) and (b) 0.01 mol/L Tris-HCl (pH 8.2) containing 0.05 mol/L AAP; (iv) photocurrent responses of ITO/Ag2S/Bi2S3/Au-ALP in (a) 0.01 mol/L Tris-HCl (pH 8.2) containing 0.05 mol/L AAP and (b) 0.01 mol/L Tris-HCl (pH 8.2) containing 0.05 mol/L AAP in the presence of 50 nmol/L NaF[105]. Copyright 2024, American Chemical Society |
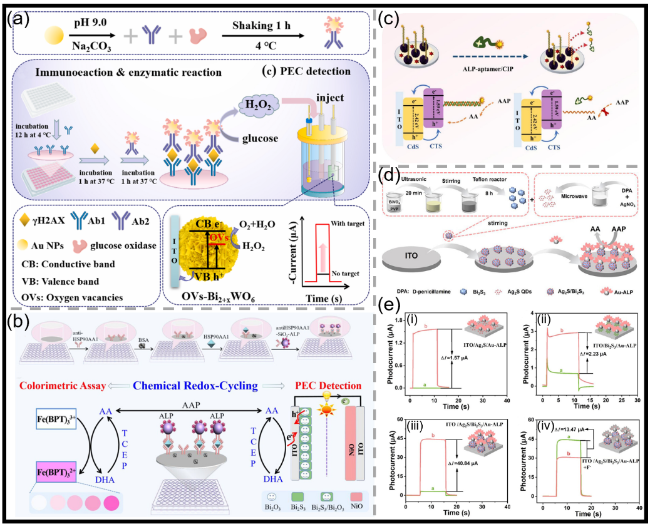
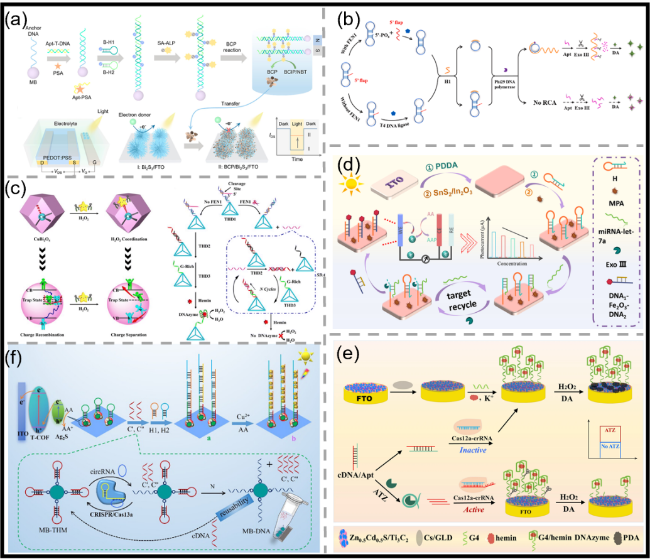
图11 (a)HCR增强的生物催化沉淀(BCP)门控OPECT适配体传感器示意图,以及基于PSA触发的HCR及随后的ALP驱动的BCP反应原理(结合磁分离辅助),所构建的OPECT适配体传感器及其基于BCP门控效应的工作原理示意图[116];(b)FEN1检测的PEC分析示意图[124];(c)H2O2在CuBi2O4纳米多面体上的自配位用于表面陷阱态修复及增强电荷分离的示意图,以及基于四面体DNA辅助链置换扩增反应的FEN1检测流程示意图[129];(d)传感器构建流程示意图[136];(e)基于CRISPR/Cas12a G4/hemin DNAzyme级联催化信号放大的PEC适配体传感器用于ATZ检测的示意图[144];(f)靶标激活的CRISPR/Cas13a反式切割三螺旋分子结构示意图,以及基于CRISPR/Cas13a编程的Cu NCs与Z型T-COF/Ag2S异质结的circRNA检测PEC生物传感器构建示意图[145]Fig.11 (a) Schematic of the HCR-enhanced biocatalytic precipitation (BCP)-gated OPECT aptasensor, and principle of PSA-dependent HCR and subsequent ALP-enabled BCP reaction with the assistance of magnetic separation. Construction of the proposed OPECT aptasensor and its working principle based on the gating effect of BCP[116]. Copyright 2023, American Chemical Society. (b) Diagram of PEC assay for FEN1 detection[124]. Copyright 2024, Elsevier. (c) Schematic illustration of the self-coordination of H2O2 onto CuBi2O4 nanopolyhedra for surface trap state remediation and reinforced charge carrier separation, and the bioassay processes for probing FEN1 with the THD-assisted SDA reaction[129]. Copyright 2023, The Royal Society of Chemistry. (d) Diagram of the process of building a sensor[136]. Copyright 2025, Elsevier. (e) Schematic of PEC aptamer sensor based on signal amplification by cascade catalysis of CRISPR/Cas12a and G4/hemin DNAzyme for ATZ detection[144]. Copyright 2025, Springer Nature. (f) Diagram of target-activated CRISPR/Cas13a trans-cleavage triple-helix molecular, and construction diagram of pec biosensor for circRNA detection CRISPR/Cas13a-programmed Cu NCs and Z-scheme T-COF/Ag2S[145]. Copyright 2025, American Chemical Society |
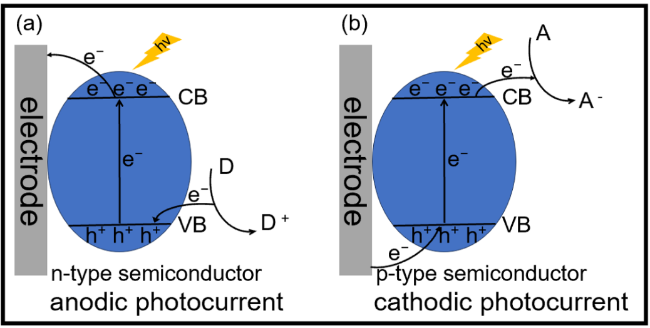
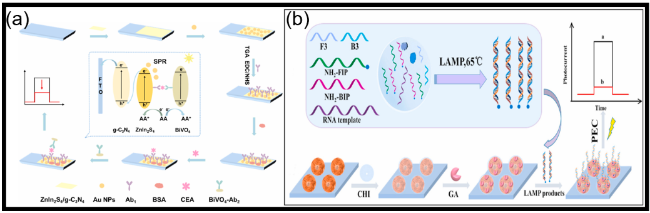
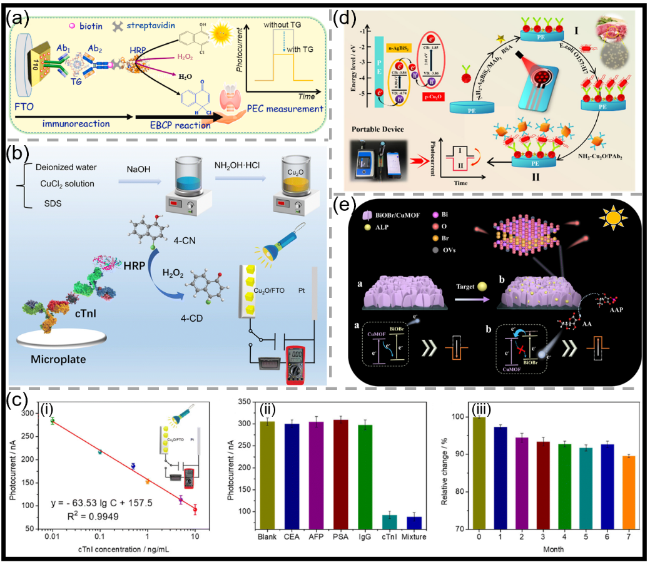
图12 (a)基于纳米金功能化BiVO4(AuNPsBiVO4)光阳极结合酶促生物催化沉淀(EBCP)的TG PEC免疫分析示意图[149];(b)基于Cu2O修饰的Cu2O/FTO电极结合EBCP反应与数字万用表读出的分裂型PEC免疫分析用于检测心肌肌钙蛋白I(cTnI)的示意图(4-CD:苯并-4-氯己二烯酮;SDS:十二烷基硫酸钠)[150];(c)(i)基于Cu2O/FTO PEC免疫分析的光电流峰值强度随cTnI浓度(0.01~10 ng/mL)变化的校准曲线;(ii)基于Cu2O/FTO PEC免疫分析的特异性及(iii)储存稳定性(特异性和稳定性测试中使用10 ng/mL的cTnI)[150];(d)用于大肠杆菌O157:H7检测的近红外响应型光电流极性可切换PEC免疫分析示意图[163];(e)用于ALP测定的光电流极性切换型PEC分析示意图[164]Fig.12 (a) Schematic illustration of PEC immunoassay for TG on nanogold-functionalized BiVO4 (AuNPsBiVO4) photoanode coupling with enzymatic biocatalytic precipitation (EBCP)[149]. Copyright 2023, Elsevier. (b) Schematic illustration of a split-type PEC immunoassay toward the detection of cardiac troponin I (cTnI) on a Cu2O modified Cu2O/FTO electrode by coupling with an EBCP reaction with a digital multimeter readout (4-CD: benzo-4-chlorohexadienone; SDS: sodium dodecyl sulfate)[150]. Copyright 2023, Royal Society of Chemistry. (c) (i) Calibration plots corresponding to the photocurrent peak intensity of the Cu2O/FTO-based PEC immunoassay as a function of cTnI concentration (0.01~10 ng/mL); (ii) the specificity and (iii) the storage stability of the Cu2O/FTO-based PEC immunoassay (10 ng/mL of cTnI for specificity and stability)[150]. Copyright 2023, Royal Society of Chemistry. (d) Schematic illustration of an NIR-responsive photocurrent-polarity-switchable PEC immunoassay for E. coli O157:H7[163]. Copyright 2023, American Chemical Society. (e) Schematic diagram of photocurrent-polarity switching PEC for ALP determination[164]. Copyright 2023, American Chemical Society |
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|
| [24] |
|
| [25] |
|
| [26] |
|
| [27] |
|
| [28] |
|
| [29] |
|
| [30] |
|
| [31] |
|
| [32] |
|
| [33] |
|
| [34] |
|
| [35] |
|
| [36] |
|
| [37] |
|
| [38] |
|
| [39] |
|
| [40] |
|
| [41] |
|
| [42] |
|
| [43] |
|
| [44] |
|
| [45] |
|
| [46] |
|
| [47] |
|
| [48] |
|
| [49] |
|
| [50] |
|
| [51] |
|
| [52] |
|
| [53] |
|
| [54] |
|
| [55] |
|
| [56] |
|
| [57] |
|
| [58] |
|
| [59] |
|
| [60] |
|
| [61] |
|
| [62] |
|
| [63] |
|
| [64] |
|
| [65] |
|
| [66] |
|
| [67] |
|
| [68] |
|
| [69] |
|
| [70] |
|
| [71] |
|
| [72] |
|
| [73] |
|
| [74] |
|
| [75] |
|
| [76] |
|
| [77] |
|
| [78] |
|
| [79] |
|
| [80] |
|
| [81] |
|
| [82] |
|
| [83] |
|
| [84] |
|
| [85] |
|
| [86] |
|
| [87] |
|
| [88] |
|
| [89] |
|
| [90] |
|
| [91] |
|
| [92] |
|
| [93] |
|
| [94] |
|
| [95] |
|
| [96] |
|
| [97] |
|
| [98] |
|
| [99] |
|
| [100] |
|
| [101] |
|
| [102] |
|
| [103] |
|
| [104] |
|
| [105] |
|
| [106] |
|
| [107] |
|
| [108] |
|
| [109] |
|
| [110] |
|
| [111] |
|
| [112] |
|
| [113] |
|
| [114] |
|
| [115] |
|
| [116] |
|
| [117] |
|
| [118] |
|
| [119] |
|
| [120] |
|
| [121] |
|
| [122] |
|
| [123] |
|
| [124] |
|
| [125] |
|
| [126] |
|
| [127] |
|
| [128] |
|
| [129] |
|
| [130] |
|
| [131] |
|
| [132] |
|
| [133] |
|
| [134] |
|
| [135] |
|
| [136] |
|
| [137] |
|
| [138] |
|
| [139] |
|
| [140] |
|
| [141] |
|
| [142] |
|
| [143] |
|
| [144] |
|
| [145] |
|
| [146] |
|
| [147] |
|
| [148] |
|
| [149] |
|
| [150] |
|
| [151] |
|
| [152] |
|
| [153] |
|
| [154] |
|
| [155] |
|
| [156] |
|
| [157] |
|
| [158] |
|
| [159] |
|
| [160] |
|
| [161] |
|
| [162] |
|
| [163] |
|
| [164] |
|
| [165] |
|
| [166] |
|
| [167] |
|
| [168] |
|
/
| 〈 |
|
〉 |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}